
Abstract
Arbuscular mycorrhizal (AM) fungi may help protect plants against herbivores; however, their use for the pest control of woody plants requires further study. Here, we investigated the effect of Glomus mosseae colonization on the interactions between gypsy moth larvae and Populus alba × P. berolinensis seedlings and deciphered the regulatory mechanisms underlying the mycorrhizal-induced resistance in the leaves of mycorrhizal poplar using RNA-seq and nontargeted metabolomics. The resistance assay showed that AM fungus inoculation protected poplar seedlings against gypsy moth larvae, as evidenced by the decreased larval growth and reduced larval survival. A transcriptome analysis revealed that differentially expressed genes (DEGs) were involved in jasmonic acid biosynthesis (lipoxygenase, hydroperoxide dehydratase, and allene oxide cyclase) and signal transduction (jasmonate-ZIM domain and transcription factor MYC2) and identified the genes that were upregulated in mycorrhizal seedlings. Except for chalcone synthase and anthocyanidin synthase, which were downregulated in mycorrhizal seedlings, all DEGs related to flavonoid biosynthesis were upregulated, including 4-coumarate-CoA ligase, chalcone isomerase, flavanone 3-hydroxylase, flavonol synthase, and leucoanthocyanidin reductase. The metabolome analysis showed that several metabolites with insecticidal properties, including coumarin, stachydrine, artocarpin, norizalpinin, abietic acid, 6-formylumbelliferone, and vanillic acid, were significantly accumulated in the mycorrhizal seedlings. These findings suggest the potential of mycorrhiza-induced resistance for use in pest management of woody plants and demonstrate that the priming of JA-dependent responses in poplar seedlings contributes to mycorrhiza-induced resistance to insect pests.
Similar content being viewed by others


Introduction
Arbuscular mycorrhizal (AM) fungi (AMF) that belong to the subphylum Glomeromycotina occur in soil and are widely distributed in agroforestry ecosystems. Fossil records and molecular data show that AMF coevolved with terrestrial plants and their symbiotic relationship was established 450 million years ago1. Arbuscular mycorrhizae represent the most common form of symbiosis between AMF and host plants, with associations observed with ~80% of terrestrial vascular plants2. AMF hyphae typically penetrate the root epidermis to colonize cortical cells and then form arbuscules composed of highly branched hyphae ensheathed in a plant-derived membrane termed the periarbuscular membrane3,4. Through AM symbiosis, AMF can transfer absorbed water and mineral nutrients (e.g., phosphate and nitrogen) to the host plant; in exchange, the host plant transports carbohydrates synthesized by photosynthesis to AMF in the form of hexose, which is used as the carbon source of fungi5. This mutualistic association promotes material exchange and information transmission between AMF and host plants, thereby achieving an optimal symbiotic state by effectively controlling the nutrient balance between the host and the fungi3,5. Intriguingly, AM symbiosis triggers a systemic effect on underground and epigeous portions of plants beyond nutritional benefits, including improving photosynthetic processes, promoting changes in primary and secondary metabolism, enhancing reactive oxygen scavenging ability, and maintaining water uptake, osmotic potential and cell membrane integrity6,7. These alterations imply that root colonization by AMF not only affects plant growth but can also regulate plant responses to biotic (e.g., bacterial pathogens and nematodes) or abiotic stress (e.g., climatic changes, water deficit, and heavy metal pollution)8.
During their lengthy interaction with phytophagous insects, plants have evolved defense mechanisms to reduce herbivore damage. Plant resistance to insects can be divided into constitutive resistance and inducible resistance. Inducible resistance is characterized by its broad spectrum and rapid effects; moreover, it can enhance the defense ability of plants against insect attacks when stimulated by specific factors9,10. Currently, inducible resistance is a research hotspot in phytochemical ecology. Many studies have demonstrated that inducible resistance in plants can be triggered by biotic stimuli, such as nonpathogenic microbial infection and pest feeding, and abiotic stimuli, such as mechanical damage and hormone treatment11,12. Studies on mycorrhizae have shown that AMF inoculation, as a biotic stimulus, can also activate inducible resistance and protect plants against herbivorous insects and bacteria13,14,15. For example, Schoenherr et al.16 reported that root colonization by the AM fungus Rhizophagus irregularis improved potato resistance to the cabbage looper (Trichoplusia ni), as evidenced by decreases in the weight gain of larvae that fed on mycorrhizal plants compared with nonmycorrhizal plants. Further research has led to the proposal of the “mycorrhizal-induced resistance” hypothesis to explain the boost in basal defenses in mycorrhizal plants17. Several studies have revealed that mycorrhizal-induced resistance appears to be a cumulative effect of mycorrhizal colonization on the plant that involves alteration of plant morphology, activation of the jasmonic acid (JA) signaling pathway, and biosynthesis of defense metabolites (e.g., flavonoids)18,19. With regard to its cost-effective and sustainable solution for plant pest control, mycorrhizal symbiosis has been regarded as an alternative to chemical pesticides and fertilizers in sustainable agricultural practices. However, positive or neutral effects of mycorrhizal symbiosis on the performance of phytophagous insects, especially specialist-chewing insects, have also been observed in several mycorrhizal plants20,21,22. Thus, it appears that the outcome of the mycorrhizal plant-herbivore interaction may vary with the AMF, host plant, and insect species involved as well as environmental factors.
Since root colonization by AMF can modify the pairwise interactions between plants and phytophagous insects, it would be useful to expand the application potential of mycorrhizal-induced resistance in plant protection and to better understand the mechanism by which AMF affect plant resistance to insects. There are many mycorrhiza-related studies that have focused on plants, such as rice, tomato, potato, wheat, and sorghum sudangrass15,16,23,24. However, these studies mainly focused on cultivated crops, and less information has been obtained on the tripartite interaction involving an AM fungus, a woody plant and a phytophagous insect. Furthermore, little is known about the systemic transcriptome and metabolome phenotypes in mycorrhizal plants that affect plant interactions with aboveground herbivores.
Populus alba × P. berolinensis is widely distributed in Northeast China, and it is an excellent tree species for fast-growing and high-yield plantations and a preferred species for shelter-belt forests, water conservation forests, and urban ornamental trees. The Asian gypsy moth (Lymantria dispar) is an economically important insect pest in the Asia-Pacific region that can also be used as an excellent biological indicator for assessing plant resistance to insects due to its well-known developmental stages and short life cycle25. In the present study, we investigated whether inoculation with a commercial inoculum containing a single Glomus mossae strain affects poplar (P. alba × P. berolinensis) seedling growth and its resistance to gypsy moth larvae. Furthermore, we integrated transcriptome and metabolomic analyses of mycorrhizal plant leaf tissues to determine the regulatory mechanism underlying the effect of AM symbiosis on poplar resistance to gypsy moth larvae at the translational and metabolic levels. Using this factorial design, we examined the following possibilities: (1) colonization of roots by AM fungus can promote generalist-chewing insect resistance in poplar seedlings; (2) priming of JA-dependent responses through the JA biosynthesis and signal transduction pathways as well as the flavonoid biosynthesis pathway may promote mycorrhizal-induced insect resistance in poplar seedlings. Relevant findings in this study could improve our understanding of the tripartite interaction among woody plants, generalist-chewing insects, and AM fungi and enhance the application potential of “mycorrhiza-induced resistance” for pest management of woody plants.
Results
AM fungal root colonization and growth evaluation of poplar seedlings
A colonization analysis revealed that the commercial mycorrhizal inoculum G. mosseae succeeded in establishing a mutualistic association with P. alba × P. berolinensis seedlings, with a 72.46% mycorrhizal infection rate in the GM group (Fig. S1A and B). Compared to the nonmycorrhizal plants, AM symbiosis in the poplar seedlings significantly improved the root length and root fresh/dry weights but had no significant effects on the plant height or the fresh/dry weights of the aboveground parts (Table S1).
Resistance of mycorrhizal poplar plants to gypsy moth larvae
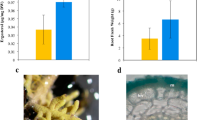
To study the resistance of woody plants that were colonized with AM fungus to insects, we analyzed the growth and survival of gypsy moth larvae that fed on the leaves of P. alba × P. berolinensis seedlings in which the roots were colonized with G. mosseae or control leaves from plants without mycorrhizal colonization. Compared to the control, the AM fungus treatment significantly decreased the body weights/lengths and head capsule widths of the 4th and 5th instar gypsy moth larvae and prolonged the development of the 3rd–5th instar larvae (Fig. 1A–D). The larval survival rate in the GM group was significantly lower than that in the CK group, as shown in the survival curve (Fig. 1E and Table S2).

Larval body weight (A). Larval body length (B). Larval head capsule width (C). Larval developmental duration (D). Larval survival curve (E). Values in the growth parameters are given as the means + standard deviations (n = 24). Asterisks above the bars indicate significant differences among groups (for (A–D), t test, n = 24; for E, log-rank test, n = 80): **P < 0.01. GM Glomus mossae-treated group, CK untreated group.
Overview of the RNA-seq and functional analyses of DEGs
To decipher the resistance response of P. alba × P. berolinensis seedlings to gypsy moth larvae at the molecular level that were affected by AM symbiosis, genome-wide transcript abundances in the seedling leaves were compared between the GM group and the CK group. A total of 127,960 transcripts and 46,397 genes were obtained from the six leaf samples using the Illumina HiSeq High-throughput sequencing platform (Illumina, USA), with average lengths of 1057 bp and 957 bp (ranging from 201 bp to 16491 bp) and N50 values of 1566 bp and 1597 bp, respectively (Table S3). A quality analysis of the transcriptome sequencing showed that mycorrhizal and nonmycorrhizal plants had average GC (%) contents of 45.72% and 45.76% clean reads, Q20 contents of 98.26% and 98.31%, and Q30 contents of 94.46% and 94.51%, respectively (Table S3). To acquire the overall genetic information, all unigenes were BLASTX searched against six databases, including the KEGG (Kyoto Encyclopedia of Genes and Genomes), eggNOG (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups), Swiss-Prot (Swiss-Prot protein sequence database), Pfam (Protein family), Nr (Nonredundant protein sequences) and GO (Gene Ontology) databases. The results showed that there were 46397 annotated genes, of which 32.26% were annotated by all six databases and 42.40% matched to at least one public database (Fig. S2). Statistical assessment of differentially expressed unigenes between the GM treatment and CK groups revealed that a total of 3671 genes (FDR < 0.05 and |log2FC|> 1; ca. 7.9% of all genes) expressed in the poplar leaves showed differential expression under AM fungus inoculation, including 2427 up- and 1244 down-regulated DEGs (Fig. S3A/B). To further validate the transcriptome data, 12 randomly selected DEGs were analyzed by RT-PCR, and they presented similar expression patterns as those observed in the RNA-seq results (Fig. S4).
To gain insights into the functional categories that differed between the GM and CK groups, GO and KEGG pathway analyses of these DEGs were performed. The GO enrichment analysis classified 3005 DEGs into 2310 terms related to biological processes, cellular components and molecular function, with 368 significantly enriched GO terms (P < 0.05). Of these, photosynthesis was the most enriched category annotated for biological processes (Table S4). Thylakoid, chloroplast stroma and chloroplast thylakoid were the major categories annotated under cellular components (Table S4). For the GO terms of the molecular function, the major categories included hydrolase activity, hydrolyzing O-glycosyl compounds and chlorophyll binding (Table S4). The KEGG analysis also assigned 2217 DEGs to 128 pathways (Fig. S5). Of these, 77 were significantly enriched KEGG pathways (P < 0.05). Further classification of the KEGG pathways showed that metabolic pathways (e.g., photosynthesis and glyoxylate and dicarboxylate metabolism) accounted for 90.91% of these significantly enriched pathways and contained nine secondary classifications that were composed of carbohydrate metabolism, energy metabolism, lipid metabolism, amino acid metabolism, other amino acid metabolism, glycan biosynthesis and metabolism, metabolism of cofactors and vitamins, terpenoid and polyketide metabolism, and other secondary metabolite biosynthesis.
Photosynthesis-related DEGs induced by AM symbiosis in leaves
A total of 130 genes associated with photosynthesis were identified by transcriptome sequencing. Among these, 97 DEGs were considered to be responsive to AM symbiosis in mycorrhizal poplar leaves and were enriched in the following three KEGG pathways: 43 DEGs belonging to photosynthesis, 38 DEGs for carbon fixation in photosynthetic organisms and 16 DEGs belonging to photosynthesis-antenna proteins (Fig. 2). All DEGs related to photosynthesis and photosynthesis-antenna proteins were upregulated, although in the carbon fixation pathway, most DEGs (e.g., NADP-dependent malic enzyme and aspartate aminotransferase) related to the C4-dicarboxylic acid cycle were downregulated while most reductive pentose phosphate cycle-related DEGs were upregulated (Fig. 2).

GM Glomus mossae-treated group, CK untreated group. Heatmap analysis was performed using the OmicStudio tools (https://www.omicstudio.cn/tool).
Transcription factors involved in the defense of mycorrhizal poplar plants
Given the important regulatory functions of transcription factors (TFs), TF-encoding genes were analyzed by sequence alignment with the Plant Transcription Factor Database (http://plntfdb.bio.uni-potsdam.de/v3.0/). A total of 90 DEGs (44 up- and 46 down-regulated) were identified as TFs, and they were distributed in 14 families and accounted for 5.7% of all the DEGs regulated by AM symbiosis. Of all the differentially expressed TFs, the ERF family showed the largest percentage (~27.8%; 25 DEGs), followed by bHLH (18 DEGs), WRKY (13 DEGs), MYB (11 DEGs), NF-YA (4 DEGs), GATA (3 DEGs), NAC (3 DEGs), Trihelix (3 DEGs), and HSF (3 DEGs). DEGs annotated as members of the NF-YA and GATA families were all upregulated. Most of the AM fungus-responsive TFs belonging to the MYB, BHLH NAC and Trihelix families were upregulated, whereas more than half of the identified TFs from the ERF, WRKY, BZIP, and HSF families were downregulated.
Identification of DEGs associated with mycorrhizal-induced resistance
To focus on how AM symbiosis influences defense responses related to mycorrhizal-induced resistance, the expression levels of the most likely gene candidates in the jasmonic acid (JA) signaling pathway and flavonoid biosynthesis were depicted. An expression analysis of the CK and GM groups identified a total of six JA signaling pathway-related genes that were responsive to the G. mossae treatment, with four genes involved in JA biosynthesis and two genes involved in JA signal transduction (Fig. 3). Among these genes, OPC8 CoA ligase (OPCL) was significantly downregulated while the remaining genes were upregulated in mycorrhizal poplar plants, including lipoxygenase (LOX), hydroperoxide dehydratase (AOS), allene oxide cyclase (AOC), jasmonate-ZIM domain (JAZ), and transcription factor MYC2 (MYC2) (Fig. 3). One gene involved in the formation of cinnamoyl-CoA (an initial metabolite in flavonoid biosynthesis) and six genes involved in flavonoid biosynthesis were identified to be responsive to the AM fungus treatment (Fig. 4). Of these, chalcone synthase (CHS) and anthocyanidin synthase (ANS) genes were significantly downregulated while 4-coumarate-CoA ligase (4CL), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), flavonol synthase (FLS), and leucoanthocyanidin reductase (LAR) genes were significantly upregulated upon AM symbiosis (Fig. 4).

The orange and gray boxes indicate upregulated and downregulated genes, respectively. Lipoxygenase (LOX); 13(S)-hydroperoxylinolenic acid (13(S)-HPOT); hydroperoxide dehydratase (AOS); 12,13(S)-epoxylinolenic acid (12,13(S)-EOT); allene oxide cyclase (AOC); 12-oxo-phytodienoic acid (12-OPDA); 3-oxo-2-(2′ (Z)-pentenyl)-cyclopentane-1-octanoic acid (OPC8); OPC8 CoA ligase (OPCL); jasmonoyl-L-isoleucine (JA-Ile); jasmonate-ZIM domain (JAZ); transcription factor MYC2 (MYC2). GM Glomus mossae-treated group, CK untreated group. Heatmap analysis was performed using the OmicStudio tools (https://www.omicstudio.cn/tool).

The orange and gray boxes indicate that genes are upregulated and downregulated, respectively. Phenylalanine ammonia-lyase (PAL); 4-coumarate-CoA ligase (4CL); cinnamic acid-4-hydroxylase (C4H); chalcone synthase (CHS); chalcone isomerase (CHI); isoflavone synthase (IFS); flavone synthase (FNS); flavanone 3-hydroxylase (F3H); flavonol synthase (FLS); leucoanthocyanidin reductase (LAR); anthocyanidin synthase (ANS). GM Glomus mossae-treated group. CK untreated group. Heatmap analysis was performed using the OmicStudio tools (https://www.omicstudio.cn/tool).
Metabolite analysis of mycorrhizal poplar leaves
To analyze the variation in metabolomic profiles in response to AM symbiosis, a nontargeted metabolomics analysis of poplar leaf samples was conducted via UPLC-QTOF MS in ESI+ mode. A total of 603 metabolites were successfully identified in both nonmycorrhizal and mycorrhizal poplar leaves and could be classified into 58 major classes. Among these metabolites, “flavonoids” (52), “benzene and substituted derivatives” (51), “glycerophospholipids” (40), “organooxygen compounds” (33), “prenol lipids” (27), and “fatty acyls” (24) were the top six accumulated metabolite groups (Fig. S6). A PCA (principal component analysis) of the resulting metabolite data revealed a considerable separation of the GM group from the CK group, with 47.75% and 6.10% variations explained by the first two principal components (PC1 and PC2, respectively) (Fig. S7A). Score plots from the PLS-DA (supervised partial least squared discriminant analysis) model also showed a clear separation between the GM group and the CK group (Fig. S7B). The model parameter R2 and Q2 intercept values analyzed after 200 permutation tests were 0.996 and −0.643, respectively, suggesting that the robustness of the models presented low overfitting and reliability risks (Fig. S7C).
By filtering with VIP > 1 and a P value < 0.05, discriminant analyses identified up to 74 metabolites that were differentially regulated in the leaves of mycorrhizal poplar seedlings compared with the nonmycorrhizal control poplar seedlings (Fig. 5A). Of these metabolites, the contents of 48 and 26 metabolites were reduced and enriched in the GM group, respectively. The most significantly altered metabolites between the mycorrhizal and nonmycorrhizal plants were carboxylic acids and derivatives and flavonoid compounds. Further in-depth comparative analysis indicated that a majority of metabolites associated with resistance against insects, including flavonoids (e.g., catechin, procyanidin B1, procyanidin B2, proanthocyanidin A2, and epicatechin), carboxylic acid derivatives (e.g., betaine), cinnamic acid derivatives (e.g., 3-hydroxycinnamic acid), and coumarin derivatives (e.g., gerberinol), were decreased in the leaves of the mycorrhizal poplar plants (Fig. 5B). However, the content of several metabolites with insecticidal properties, including coumarin, stachydrine, artocarpin, norizalpinin, abietic acid, 6-formylumbelliferone, and vanillic acid, was significantly increased in the leaves of mycorrhizal poplar plants (Fig. 5B). Through metabolic pathway mapping with the MetaboAnalyst website (https://www.metaboanalyst.ca/), these differentially expressed metabolites were enriched in 42 KEGG pathways (Fig. S8). On the basis of both the lg(P value) and pathway impact scores, the most relevant metabolic pathways in response to AM symbiosis were isoquinoline alkaloid biosynthesis, flavone and flavonol biosynthesis, glycerolipid metabolism, phenylalanine metabolism, and glycerophospholipid metabolism (Fig. S8).

A Heatmap analysis of 74 differentially expressed metabolites between nonmycorrhizal (CK) and mycorrhizal (GM) plants. The figure shows the data processed by the log2 function and mean subtraction in turn. B Relative abundance of differentially expressed metabolites with insecticidal properties. Heatmap analysis was performed using the OmicStudio tools (https://www.omicstudio.cn/tool).
Discussion
AM symbiosis, as a potential broad-spectrum and effective pest control strategy, may represent a valid alternative to some chemical pesticides in contemporary agriculture26. Based on the “mycorrhiza-induced resistance” theory, many commercial AMF products have been developed to improve crop resistance or tolerance to insect pests27. However, the applicability of this insect resistance mechanism in woody plants is lacking. In the present study, we investigated the outcome of insect–plant–microbe interactions with a focus on P. alba × P. berolinensis seedlings, gypsy moth larvae, and G. mossae. This is the first report on the tripartite interaction among a Populus plant, an AM fungus and a generalist-chewing insect. In addition, we tracked the transcriptomic and metabolomic alterations in the leaves of the mycorrhizal and nonmycorrhizal poplar seedlings using RNA-seq and nontargeted metabolomics technologies, which provided us with an opportunity to examine the candidate genes and metabolites related to metabolic pathways underlying the resistance to insects in AM fungus-colonized woody plants.
Growth evaluation
Augmented plant growth and biomass (e.g., rice and wheat) in response to inoculation with AMF have been repeatedly reported in various laboratory or field experiments, a phenomenon generally identified as a positive mycorrhizal growth response (MGR)28,29. Positive MGRs appear to be the consequence of improved uptake and translocation of soil nutrients, generally phosphorus and nitrogen, as the hyphal network can extend the soil area of the mycorrhizal roots to absorb nutrients24,30. However, in the present study, under the AM fungus treatment, although the biomass of root tissues was significantly increased, the plant height as well as the fresh and dry weights of the aboveground parts were not significantly different from those of the nonmycorrhizal poplar seedlings. These results clearly suggested a species- or tissue-specific growth response to AM fungi in plants.
The transcriptomic analysis showed that AM symbiosis significantly activated photosynthesis-related pathways in the leaves of the P. alba × P. berolinensis seedlings. All photosynthesis and photosynthesis antenna protein-related DEGs and most of the reductive pentose phosphate cycle-related DEGs were upregulated. Previous studies demonstrated that during the symbiosis between AMF and plants, AMF competed with the host plants for photosynthates and the carbon consumed by AMF could account for up to 4–20% of photosynthetically assimilated carbon26,31. Our results along with the findings of previous studies unambiguously reveal that improved photosynthetic efficiency in plants is a major regulatory mechanism for maintaining a mutualistic association with AMF. In addition, photosynthesis is the most critical factor restricting plant growth, and its improvement usually promotes plant growth. Nevertheless, we observed a neutral MGR for the aboveground biomass (growth) of P. alba × P. berolinensis seedlings in this study. Therefore, based on the consumption of photosynthates by mycorrhizal colonization, we believe that the neutral MGR may be attributed to the possible negative effects of carbon losses in the host plant caused by AMF inoculation reaching a balance with the benefits in growth acquired from the increased nutrient supply or activated photosynthesis. Notably, growth inhibition in mycorrhizal plants (negative MGR) might even be triggered when the cost of carbon losses to the plant exceeds the benefit in growth acquired from increased nutrient supply. This situation has been previously reported in some cultivated crops32,33.
Resistance response to insects
Systemic protection by mycorrhizal symbiosis can also be found in the aerial parts of AMF-inoculated plants, although the consequence of mycorrhizal fungus-plant-herbivore interactions is variable and may have a positive, negative, or mixed effect18,34. Consistent with previous studies in crops, such as alfalfa, rice, wheat, and tomato15,23,35,36, our results showed that treating poplar seedlings with AM fungus significantly decreased the survival, body weight, body length, and head capsule width of gypsy moth larvae and significantly prolonged the duration of larval development. This finding indicates that AM fungal colonization increased the resistance of P. alba × P. berolinensis seedlings to gypsy moth larvae. The present study provides evidence that AM symbiosis can lead to priming in plant tissues for a more rapid defensive response and that mycorrhiza-induced resistance is also applicable to woody plants. It is worth mentioning that the larval survival rate in the GM group remained high while the development duration was greatly increased. Prolonged development of gypsy moth larvae under unfavorable conditions has been reported in several studies, such as under heavy metal stress37,38. These results indicate that delaying development might be an effective survival strategy for gypsy moth larvae to obtain sufficient energy for growth under adverse environments.
Defense response in mycorrhizal poplar seedlings
AMF inoculation can elicit inducible resistance in plants by activating a broad range of defense genes that are expressed only after pathogen or insect attack16,17. The key regulators of mycorrhiza-induced resistance are plant hormones, such as jasmonic acid and salicylic acid, which usually accumulate in plants with AMF colonization18,26. The JA signaling pathway is an important defense response in many plants39; therefore, we systematically elaborated the expression model of genes involved in JA biosynthesis and JA signal transduction in mycorrhizal poplar seedlings. Our results showed that three JA biosynthesis-related genes (LOX2, AOS, and AOC) and two JA signal transduction-related genes (JAZ and MYC2) were upregulated in mycorrhizal seedlings compared to nonmycorrhizal seedlings, and they represent key regulatory genes in the JA signaling pathway. For example, under the continuous catalysis of LOX, AOS, and AOC, alpha-linolenic acid can be transformed into the JA precursor 12-oxo-phytodienoic acid40. Therefore, their upregulation at the mRNA level indicates that AM fungus inoculation stimulates the JA signaling pathway in the leaves of P. alba × P. berolinensis seedlings and demonstrates that activation of the JA signaling pathway is a major component of mycorrhiza-induced resistance. Similar results were reported by Schoenherr et al.16, who observed systemic upregulation of the transcript levels of AOS1 and 12-oxo-phytodienoate reductase 3 (belonging to the JA signaling pathway) in potatoes that were colonized with the AM fungus Rhizophagus irregularis.
Flavonoids have insecticidal effects and help protect plants against phytophagous insects41,42. The biosynthesis of flavonoid compounds is generally divided into the following three stages: L-phenylalanine to 4-coumaroyl-CoA, which is catalyzed by PAL, 4CL and cinnamic acid-4-hydroxylase (C4H); 4-coumaroyl-CoA to flavonone, which is catalyzed by CHS and CHI; and flavonone to various types of flavonoids, including isoflavones, which are catalyzed by isoflavone synthase (IFS), flavones, which are catalyzed by flavone synthase (FNS), flavonols, which are catalyzed by FLS, flavanols, which are catalyzed by LAR, and anthoyanidins, which are catalyzed by ANS43. In the present study, a total of seven rate-limiting enzymes in the flavonoid biosynthesis pathway were significantly altered at the mRNA level in the leaves of P. alba × P. berolinensis seedlings under the AM fungus treatment, and they were distributed in the three stages mentioned above. Of these, 4CL, CHI, F3H, FLS, and LAR were upregulated, while CHS and ANS were downregulated. These results suggest that AM symbiosis may improve the biosynthesis of flavonols and flavanols and suppress the accumulation of anthocyanidins. Furthermore, TF-encoding gene analysis showed that the transcription factor families involved in flavonoid biosynthesis, including the MYB, bHLH, and NAC families44,45,46, were responsive to mycorrhizal symbiosis, and most TFs of these three families were upregulated in the AMF treatment group. Taken together, the biosynthesis of flavonoids was strongly affected by AMF colonization, which may be the most direct factor underlying the enhancement of resistance to gypsy moth larvae in P. alba × P. berolinensis seedlings under the AM fungus treatment.
Nontargeted metabolomics technology provides an opportunity to identify the metabolites involved in mycorrhiza-induced resistance in the leaves of P. alba × P. berolinensis seedlings. Consistent with the results reported by Wang et al.47, our results showed that flavonoids were the most abundant class among all the identified metabolites. A discriminant analysis revealed that not all differentially accumulated metabolites with insecticidal properties in mycorrhizal poplar plants were significantly increased. For example, catechin, betaine, and 3-hydroxycinnamic acid, which were previously shown to interfere with insect growth and survival48,49,50, were significantly decreased in the present study. However, several metabolites related to plant resistance to insects, including coumarin, stachydrine, artocarpin, norizalpinin, abietic acid, 6-formylumbelliferone, and vanillic acid, were significantly accumulated in the leaves of mycorrhizal poplar seedlings. These results suggest that AM fungus G. mosseae colonization did not improve the chemical defense of P. alba × P. berolinensis seedlings in a universal fashion. Increases in these insecticidal metabolites may be a direct defense mechanism for the inhibition of L. dispar larval growth and survival in the GM treatment group; thus, these compounds might be useful for the development of novel botanical insecticides that can control gypsy moths.
In summary, the AM fungus G. mosseae, P. alba × P. berolinensis seedlings and gypsy moth larvae were successfully employed to analyze the mycorrhiza-induced resistance of a woody plant to a generalist insect herbivore and concluded that AMF inoculation was able to shield P. alba × P. berolinensis seedlings against L. dispar larvae. Further transcriptome and metabolome analyses identified candidate genes and metabolites that are involved in mycorrhiza-induced resistance to insects in the leaves of P. alba × P. berolinensis seedlings. Special emphasis was placed on the JA signaling pathway and the biosynthesis of flavonoids and differentially accumulated metabolites with insecticidal properties. These findings help to characterize the defense responses in mycorrhizal poplar seedlings.
Materials and methods
Plant materials
The AM fungus used in this study was the mycorrhizal inoculum G. mosseae, which was purchased from the Gansu Academy of Agricultural Sciences, China. This mycorrhizal inoculum was composed of spores, mycelium, root segments and sand at a concentration of 15 propagules/g mycorrhizal inoculum. P. alba × P. berolinensis seedlings were annual cuttings purchased from a commercial market in Harbin. The soil substrate for seedlings was composed of turfy soil, vermiculite and sand in a volume ratio of 3:1:1 and was sterilized at 121 °C for 2 h in an autoclave sterilizer before planting the seedlings. In late April 2020, poplar seedlings were planted in pots (16 × 16 × 20 cm) containing a mix of 3 kg sterilized soil substrate with 20 g mycorrhizal inoculum (denoted as GM) or without GM as a noninoculated plant (denoted as CK). A total of 300 poplar seedlings (one plant/pot) were planted (n = 150 for each group) and transferred to the plastic greenhouse of Forest Genetic Breeding Nursery in Northeast Forestry University for growth.
Analysis of root colonization
Root samples of the poplar seedlings were collected 3 months after the plants were inoculated with AMF, washed with sterile water, and stained with 0.008% trypan blue as previously described13,51. Mycorrhizal colonization was evaluated under a low-power compound microscope, and the occurrence of spores or arbuscules on the root segments indicated successful colonization. Each group had four replicates, with at least 50 root segments observed for each replicate. The percent mycorrhizal infection was calculated using the following formula and analyzed using an independent sample t test (SPSS 19.0 for Windows) at α = 0.05. Mycorrhizal infection (%) = 100 × Number of infected root segments/Total number of root segments observed.
Insect assays
In March 2020, gypsy moth eggs were collected from the Forest Farm of Northeast Forestry University, Harbin, China, and stored at 4 °C before use. In early August, gypsy moth larvae hatched from the eggs, and they were subsequently reared on artificial diets (purchased from Chinese Academy of Forestry, Beijing, China) until they reached 2nd instars. Growth conditions were 25 °C, a 16:8 h (L:D) photoperiod and 60 ± 1% relative humidity in an incubator. The newly molted 2nd instar larvae were divided into two groups and maintained on leaves removed from the nonmycorrhizal and mycorrhizal poplar seedlings. Leaves were refreshed every 2 days until the larvae died or pupated. Each group contained 80 larvae, and these larvae were reared in 10 plastic boxes (18 × 8 × 6 cm). The numbers of dead and live larvae were recorded, and survival curves were generated using SPSS 19.0 software for Windows and analyzed by the log-rank test for significance. The body weight, body length, head capsule width and developmental duration of 3rd-5th instar larvae in the CK and GM groups (n = 24 in each group) were also recorded and analyzed using an independent sample t test at α = 0.05.
Transcriptome analysis
In mid-August 2020, the first five leaves from the top of each of three healthy seedlings were removed and then combined for a replicate. A total of three replicates were generated for each group (GM or CK). All poplar seedlings used for the transcriptome analysis had not been previously harvested for the rearing of gypsy moth larvae or for performing other experiments. The experimental process for the transcriptome analysis was conducted by LC-Bio Technology Co., Ltd. (Hangzhou city, China) according to the standard provided by the Illumina Company (San Diego, CA, USA), including library preparation and sequencing experiments. Total RNA from each poplar leaf sample was isolated using TRIzol reagent (Invitrogen, CA, USA) following the manufacturer’s instructions. The total RNA quantity and purity were checked using a Bioanalyzer 2100 and RNA 1000 Nano LabChip Kit (Agilent, CA, USA) with RIN number >7.0. Poly(A) RNA was purified from total RNA (5 µg) over two rounds using poly-T oligo-attached magnetic beads. After purification, the mRNA was fragmented into small pieces using divalent cations under an elevated temperature. Then, the cleaved RNA fragments were reverse-transcribed to create the final cDNA library using the mRNA Seq sample preparation kit (Illumina) in accordance with the manufacturer’s instructions, and the average insert size for the paired-end libraries was 300 bp (±50 bp). Paired-end sequencing was conducted on an Illumina HiSeq 4000 platform in accordance with the recommended protocol.
To acquire high-quality reads, in-house developed Cutadapt and Perl scripts were used to remove adapter sequences, low-quality reads and reads containing poly-N52. The sequence quality of the clean data was verified using FastQC, including the Q20, Q30, and GC content. All downstream analyses were based on clean data with high quality. De novo assembly of the transcriptome was accomplished with Trinity 2.4.0 software. All assembled unigenes were annotated using the following databases: SwissProt, GO, Nr, KEGG, and eggNOG. The expression level of each gene was analyzed using RSEM and normalized to the fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) value. Genes with an absolute value of |log2FC|> 1 and with statistical significance (P value < 0.05) using the R package edgeR were defined as differentially expressed genes (DEGs). GO and KEGG enrichment analyses of the DEGs were carried out with padj < 0.05 to identify the significantly enriched pathways.
Real-time PCR
To verify the transcriptomic data results, 12 DEGs were randomly selected and their mRNA abundances were analyzed via real-time PCR, as previously described53. In brief, cDNA was synthesized with RNA used in transcriptome analysis by the PrimeScript™ RT Reagent Kit (Takara, China), and RT-PCR was performed using the StepOnePlus™ Real-Time PCR System (Thermo) and the fluorescence dye SYBR Premix Ex TaqTM (TaKaRa, China). Each RT-PCR assay (10 µL) contained 5 µL of SYBR Premix Ex TaqTM, 0.4 µL of each primer (0.5 mmol/L), 1 µL of template cDNA, 0.2 µL of ROX Reference Dye and 3 µL of ddH2O. The amplification program included 95 °C for 30 s, followed by 95 °C for 5 s and 60 °C for 30 s (40 cycles). There were three biological replicates for each gene. The EF1β gene was used as a reference gene for normalization54, and the gene-specific primers for RT-PCR are shown in Supplementary Table S5. The relative gene expression was calculated according to the 2−△△Ct method.
Metabolome analysis
In mid-August 2020, the first five leaves from the top of each of three healthy seedlings were removed and then combined for a replicate. A total of six replicates were established for each group (GM or CK). All poplar seedlings used for metabolome analysis had not been previously harvested for rearing gypsy moth larvae or performing other experiments. The metabolome analysis was performed by LC-Bio Technology Co., Ltd. (Hangzhou city, China) using the ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry (UPLC-QTOF MS) method. In brief, poplar leaf samples were frozen in liquid nitrogen and then ground. Metabolites from the ground leaf samples were extracted with 50% methanol. One microliter of the metabolite extract was injected into an Acquity UPLC system coupled to a TripleTOF 5600 plus high‐resolution tandem mass spectrometer (SCIEX, UK) and separated with an ACQUITY UPLC T3 column (100 mm×2.1 mm, 1.8 µm, Waters, UK). LC-MS raw data files were converted into mzXML format using Proteowizard MSConver software and then processed by the XCMS, CAMERA, and metaX toolboxes implemented with R software55. The online KEGG and HMDB databases were used to annotate the metabolites by matching the exact molecular mass data (m/z) of samples with those from the database. Student’s t tests were conducted to detect differences in metabolite concentrations between the control and treatment groups. The P value was adjusted for multiple tests using an FDR (Benjamini–Hochberg Method). Multivariate data analyses, including PCA and PLS-DA, were conducted using metaX to discriminate the different variables between groups. The VIP value was calculated. Differentially expressed metabolites (DEMs) among the CK and GM groups were defined at VIP > 1 and P < 0.05.
Plant growth determination
In late August 2020, 12 unplucked poplar seedling plants in the CK or GM group were harvested and divided into roots, stems, and leaves by group. Their growth parameters, including the root length and plant height, as well as biomass parameters, including the fresh weights and dry weights of below- and aboveground parts, were recorded. Data were analyzed using an independent sample t test at α = 0.05.
Data availability
The RNA-sequencing data have been deposited in the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) database, with the accession number PRJNA753556. The other referenced data are included in the article or Supplementary Materials.
References
Smith, S. E., & Read, D. F. Mycorrhizal Symbiosis. 3rd edn. (Academic Press, 2008).
Brundrett, M. C. & Tedersoo, L. Evolutionary history of mycorrhizal symbioses and global host plant diversity. New. Phytol. 4, 1108–1115 (2018).
Wipf, D., Krajinski, F., Tuinen, D., Recorbet, G. & Courty, P. E. Trading on the arbuscular mycorrhiza market: from arbuscules to common mycorrhizal networks. New Phytol. 223, 1127–1142 (2019).
Pumplin, N. & Harrison, M. J. Live-cell imaging reveals periarbuscular membrane domains and organelle location in Medicago truncatula roots during arbuscular mycorrhizal symbiosis. Plant Physiol. 151, 809–819 (2019).
Wang, W. X. et al. Nutrient exchange and regulation in arbuscular mycorrhizal symbiosis. Mol. Plant 10, 1147–1158 (2017).
Begum, N. et al. Role of arbuscular mycorrhizal fungi in plant growth regulation: implications in abiotic stress tolerance. Front. Plant Sci. 10, 1068 (2019).
Zhou, S., Lou, Y. R., Tzin, V. & Jander, G. Alteration of plant primary metabolism in response to insect herbivory. Plant. Physiol. 169, 1488–1498 (2015).
Diagne, N., Ngom, M., Djighaly, P. I., Fall, D. & Svistoonoff, S. Roles of arbuscular mycorrhizal fungi on plant growth and performance: Importance in biotic and abiotic stressed regulation. Diversity 12, 370 (2020).
Kawazu, K., Mochizuki, A., Sugeno, W., Seo, S. & Mitsuhara, I. Differences in the susceptibility of five herbivore species and developmental stages to tomato resistance induced by methyl jasmonate treatment. Arthropod Plant Interact. 7, 415–422 (2013).
Mauch-Mani, B., Baccelli, I., Luna, E. & Flors, V. Defense priming: an adaptive part of induced resistance. Annu. Rev. Plant Biol. 68, 485–512 (2017).
Jiang, D. & Yan, S. C. MeJA is more effective than JA in inducing defense responses in Larix olgensis. Arthropod Plant Interact. 12, 49–56 (2018).
Waterman, J. M., Cazzonelli, C. I., Hartley, S. E. & Johnson, S. N. Simulated herbivory: the key to disentangling plant defence responses. Trends Ecol. Evol. 34, 447–458 (2019).
Selvaraj, A., Thangavel, K. & Uthandi, S. Arbuscular mycorrhizal fungi (Glomus intraradices) and diazotrophic bacterium (Rhizobium BMBS) primed defense in blackgram against herbivorous insect (Spodoptera litura) infestation. Microbiol. Res. 231, 126355 (2020).
Formenti, L. & Rasmann, S. Mycorrhizal fungi enhance resistance to herbivores in tomato plants with reduced jasmonic acid production. Agron.-Basel 9, 131 (2019).
Song, Y. Y. et al. Priming of anti-herbivore defense in tomato by arbuscular mycorrhizal fungus and involvement of the jasmonate pathway. Chem. Ecol. 39, 1036–1044 (2013).
Schoenherr, A. P., Eric, R., Natasha, J., Manosalva, P. & Gomez, S. K. Mycorrhiza-induced resistance in potato involves priming of defense responses against cabbage looper (Noctuidae: lepidoptera). Environ. Entomol. 48, 370–381 (2019).
Pozo, M. J. & Azcon-Aguilar, C. Unraveling mycorrhiza-induced resistance. Curr. Opin. Plant Biol. 10, 393–398 (2007).
Kadam, S. B., Pable, A. A. & Barvkar, V. T. Mycorrhiza induced resistance (MIR): a defence developed through synergistic engagement of phytohormones, metabolites and rhizosphere. Funct. Plant Biol. 47, 880–890 (2020).
Pozo, M. J., López-Ráez, J. A., Azcón-Aguilar, C. & Garcia, G. J. M. Phytohormones as integrators of environmental signals in the regulation of mycorrhizal symbioses. New Phytol. 205, 1431–1436 (2015).
Raúl, O. R. S. Increased maize growth and P uptake promoted by arbuscular mycorrhizal fungi coincide with higher foliar herbivory and larval biomass of the Fall Armyworm Spodoptera frugiperda. Mycorrhiza 29, 615–622 (2019).
Bernaola, L., Cosme, M., Schneider, R. W. & Stout, M. Belowground inoculation with arbuscular mycorrhizal fungi increases local and systemic susceptibility of rice plants to different pest organisms. Front. Plant Sci. 9, 747 (2018).
Minton, M. M., Barber, N. A. & Gordon, L. L. Effects of arbuscular mycorrhizal fungi on herbivory defense in two Solanum (Solanaceae) species. Plant. Ecol. Evol. 149, 157–164 (2016).
Bernaola, L. & Stout, M. J. The effect of mycorrhizal seed treatments on rice growth, yield, and tolerance to insect herbivores. J. Pest Sci. 10, 1–18 (2020).
Pellegrino, E., Öpik, M., Bonari, E. & Ercoli, L. Responses of wheat to arbuscular mycorrhizal fungi: a meta-analysis of field studies from 1975 to 2013. Soil Biol. Biochem. 84, 210–217 (2015).
Jiang, D., Tan, M. T., Wang, Q., Wang, G. R. & Yan, S. C. Evaluating the ecotoxicological effects of Pb contamination on the resistance against Lymantria dispar in forest plant, Larix olgensis. Pest Manag. Sci. 76, 2490–2499 (2020).
Jung, S. C., Martinez, M. A., Lopez, R. J. A. & Pozo, M. J. Mycorrhiza-induced resistance and priming of plant defenses. J. Chem. Ecol. 38, 651–664 (2012).
Igiehon, N. O. & Babalola, O. O. Biofertilizers and sustainable agriculture: exploring arbuscular mycorrhizal fungi. Appl. Microbiol. Biotechnol. 101, 4871–4881 (2017).
Zhang, S., Lehmann, A., Zheng, W., You, Z. Y. & Rillig, M. C. Arbuscular mycorrhizal fungi increase grain yields: a meta-analysis. New Phytol. 222, 543–555 (2019).
Campo, S., Héctor, M. C., Marta, O., Pla, E., & Segundo, B. S. Effect of root colonization by arbuscular mycorrhizal fungi on growth, productivity and blast resistance in rice. Rice. 13, 42 (2020).
Smith, S. E. & Smith, F. A. Roles of arbuscular mycorrhizas in plant nutrition and growth: new paradigms from cellular to ecosystem scales. Annu. Rev. Plant Boil. 62, 227–250 (2011).
Kaur, S. & Suseela, V. Unraveling arbuscular mycorrhiza-induced changes in plant primary and secondary metabolome. Metabolites 10, 335 (2020).
Verbruggen, E., Kiers, E. T., Bakelaar, P., Roling, W. F. M. & Heijden, M. G. A. Provision of contrasting ecosystem services by soil communities from different agricultural fields. Plant Soil 350, 43–55 (2012).
Grace, E. J., Cotsaftis, O., Tester, M., Smith, F. A. & Smith, S. E. Arbuscular mycorrhizal inhibition of growth in barley cannot be attributed to extent of colonization, fungal phosphorus uptake or effects on expression of plant phosphate transporter genes. New Phytol. 181, 938–949 (2009).
Miozzi, L. et al. Arbuscular mycorrhizal symbiosis: plant friend or foe in the fight against viruses? Front. Microbiol. 10, 1238 (2019).
Simon, A. L., Wellham, P. A. D., Aradottir, G. I. & Gange, A. C. Unravelling mycorrhiza-induced wheat susceptibility to the English grain aphid Sitobion avenae. Sci. Rep. 7, 46497 (2017).
Li, Y., Nan, Z. & Duan, T. Rhizophagus intraradices promotes alfalfa (Medicago sativa) defense against pea aphids (Acyrthosiphon pisum) revealed by RNA-Seq analysis. Mycorrhiza 29, 623–635 (2019).
Jiang, D. & Yan, S. C. Effects of Cd, Zn or Pb stress in Populus alba berolinensis on the development and reproduction of Lymantria dispar. Ecotoxicology 26, 1305–1313 (2017).
Borowska, J., Sulima, B., Niklinska, M. & Pyza, E. Heavy metal accumulation and its effects on development, survival and immuno-competent cells of the housefly Musca domestica from closed laboratory populations as model organism. Fresen. Environ. Bull. 13, 1402–1409 (2004).
Ruan, J., Zhou, Y., Zhou, M., Yan, J. & Zhang, K. X. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 20, 2479 (2019).
Dabrowska, P. & Boland, W. Iso-OPDA: an early precursor of cis-jasmone in plants? Chembiochem 8, 2281–2285 (2007).
Züst, T. & Agrawal, A. A. Trade-offs between plant growth and defense against insect herbivory: an emerging mechanistic synthesis. Annu. Rev. Plant Biol. 68, 513–534 (2017).
Wang, Z. Y., Nur, F. A., Ma, J. Y., Wang, J. G. & Cao, C. W. Effects of poplar secondary metabolites on performance and detoxification enzyme activity of Lymantria dispar. Comp. Biochem. Physiol. C 225, 108587 (2019).
Falcone, F. M. L., Rius, S. P. & Casati, P. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3, 222 (2012).
Xu, W. J., Dubos, C. & Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant. Sci. 20, 176–185 (2015).
Terrier, N. et al. Ectopic expression of VvMybPA2 promotes proanthocyanidin biosynthesis in Grapevine and suggests additional targets in the pathway. Plant Physiol. 149, 1028–1041 (2009).
Jiang, T. et al. Integrated transcriptomic and metabolomic data reveal the flavonoid biosynthesis metabolic pathway in Perilla frutescens (L.) leaves. Sci. Rep. 10, 16207 (2020).
Wang, Y., Liu, Q., Du, L., Hallerman, E. M. & Li, Y. H. Transcriptomic and metabolomic responses of rice plants to Cnaphalocrocis medinalis caterpillar infestation. Insects 11, 705 (2020).
Su, Q. et al. Effect of plant secondary metabolites on common cutworm, Spodoptera litura (Lepidoptera: Noctuidae). Entomol. Res. 48, 18–26 (2018).
Yang, Y. C. et al. Effects of secondary metabolites of alligator weed Alternanthera philoxeroides on the enzyme activity of amylases and glutathione S-transferases in the adults of alligator weed flea beetle Agasicles hygrophila. J. Plant. Prot. 45, 739–744 (2018).
Oszmiański, J., Kalisz, S. & Aneta, W. The content of phenolic compounds in leaf tissues of white (Aesculus hippocastanum L.) and red horse chestnut (Aesculus carea H.) colonized by the horse chestnut leaf miner (Cameraria ohridella Deschka & Dimi). Molecules 19, 14625–14636 (2014).
Phillips, J. M. & Hayman, D. Improved procedures for clearing roots and staining parasitic and vesicular-arbuscular mycorrhizal fungi for rapid assessment of infection. Trans. Brit. Mycol. Soc. 55, 158–161 (1970).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
Jiang, D. et al. Cd exposure-induced growth retardation involves in energy metabolism disorder of midgut tissues in the gypsy moth larvae. Environ. Pollut. 266, 115173 (2020).
Zhai, J. F. et al. Cloning and expression of DREB gene from Populus alba × P. berolinensis. J. Northwest A&F Univ. Nat. Sci. Ed. 6, 87–93 (2012).
Wen, B., Mei, Z., Zeng, C. & Liu, S. MetaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 18, 183 (2017).
Acknowledgements
This research was supported by the National Key Research and Development Program of China (2018YFC1200400).
Author information
Authors and Affiliations
Contributions
S.Y. and G.W. conceived the experiments, and D.J., S.W. and M.T. performed the experiments. D.J., L.Z. and Q.W. analyzed the data. D.J., S.Y. and G.W. wrote and revised the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, D., Tan, M., Wu, S. et al. Defense responses of arbuscular mycorrhizal fungus-colonized poplar seedlings against gypsy moth larvae: a multiomics study. Hortic Res 8, 245 (2021). https://doi.org/10.1038/s41438-021-00671-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00671-3
This article is cited by
-
Tree Communication: the Effects of “Wired” and “Wireless” Channels on Interactions with Herbivores
Current Forestry Reports (2022)
