
Abstract
The Dormancy-associated MADS-box (DAM) gene cluster in peach serves as a key regulatory hub on which the seasonal temperatures act and orchestrate dormancy onset and exit, chilling response and floral bud developmental pace. Yet, how different temperature regimes interact with and regulate the six linked DAM genes remains unclear. Here, we demonstrate that chilling downregulates DAM1 and DAM3–6 in dormant floral buds with distinct patterns and identify DAM4 as the most abundantly expressed one. We reveal multiple epigenetic events, with tri-methyl histone H3 lysine 27 (H3K27me3) induced by chilling specifically in DAM1 and DAM5, a 21-nt sRNA in DAM3 and a ncRNA induced in DAM4. Such induction is inversely correlated with downregulation of their cognate DAMs. We also show that the six DAMs were hypermethylated, associating with the production of 24-nt sRNAs. Hence, the chilling-responsive dynamic of the different epigenetic elements and their interactions likely define distinct expression abundance and downregulation pattern of each DAM. We further show that the expression of the five DAMs remains steadily unchanged or continuously downregulated at the ensuing warm temperature after chilling, and this state of regulation correlates with robust increase of sRNA expression, H3K27me3 and CHH methylation, which is particularly pronounced in DAM4. Such robust increase of repressive epigenetic marks may irreversibly reinforce the chilling-imposed repression of DAMs to ensure flower-developmental programming free from any residual DAM inhibition. Taken together, we reveal novel information about genetic and epigenetic regulation of the DAM cluster in peach, which will be of fundamental significance in understanding of the regulatory mechanisms underlying chilling requirement and dormancy release, and of practical application for improvement of plasticity of flower time and bud break in fruit trees to adapt changing climates.
Similar content being viewed by others

Introduction
Dormancy is an adaptation that enables perennial plants to survive unfavorable seasonal stresses. In the temperate zone where winter freezing is a major threat, plants enter the dormant state in late fall before winter to avoid freezing injury1,2. Seasonal environmental cues primarily dictate dormancy onset and development and release, which has been intensively studied in temperate perennials that undergo winter dormancy1,3,4. For example, a shortening photoperiod or declining temperature or both in the fall induce dormancy in peach (P. persica L. Batsch), while declining temperature serves as the only factor for apple (M. domestica) and pear (P. communis). In all cases, environmental cues cause the apical shoot meristem to cease growth and form a bud to enter the ecodormant state1,5. In contrast, lateral vegetative buds formed as result of apical dominant growth suppression, are in a state of paradormancy6. Both ecodormancy and paradormancy are temporary, reversible, and serve as initial stages for the transition into the deep dormant state called endodormancy in late fall5. The floral buds that initiate and form in the summer, similarly enter endodormancy near the end of fall7. Endodormancy is a physiological state that is not readily broken or released by short favorable environmental conditions unless exposed to chilling temperatures (>0–7.5 °C)1. This chilling requirement is obligatory, but varies considerably among plants, which is primarily determined by origin and genotype8. Chilling requirement also varies among different buds within the same trees or floral organs within the same flower as reflected by the longer chilling period required by the dormant floral buds compared to apical leaf buds7 and the female floral organ compared to the male organ9. Hence, the biological nature and developmental trajectory of meristems also contribute to chilling requirement.
Chilling is, in fact, essential for floral development. In contrast to dormant vegetative buds that are believed to primarily arrest at G1 phase of the cell cycle and remain quiescent during the chilling period10, the dormant peach floral buds undergo morphological changes11, with evident formation of distinct archesporial cells and epidermis, microsporangium walls and tapetum in the anthers12,13, and visible ovules in the carpel/gynoecium14,15,16. Yet, these floral morphological changes rarely occur in the fully dormant floral buds maintained at ambient or warm temperatures11. The morphological response is, in fact, chilling stage-dependent and major development events such as ovule formation in carpel only occurs near the end of the chilling period12,13,14,15,16, and insufficient chilling leads to the arrest of carpel development before or at the stage of ovule formation9. Evidently, chilling couples the dormancy release with floral developmental programming and only critical stages (e.g. ovule formation) driven through by chilling renders the floral buds capably released from dormancy or competent to grow in spring.
Warm temperature immediately following chilling plays an important role in the coordination of floral bud development and break. Even after chilling requirement is fulfilled, the floral buds still do not immediately progress to flowering unless exposed to a period of warm temperatures, a phenomenon termed “heat sum” requirement, which has been documented in numerous temperate deciduous fruit trees17,18,19. Like the chilling requirement, the heat requirement is highly heritable and often ecotypically adapted20. However, temperatures at or above 25 °C often impair floral organ development21,22. Chilling and warm requirements are interrelated and interact such that longer chilling periods lead to a shorter warm period requirement, suggesting common genes or pathways are targeted by both temperature regimes. In contrast, the warm requirement is directly related to bud break and flowering time and cannot be completely substituted by chilling10,18.
Dormancy onset and exit, and chilling and warm requirement appear to share a similar genetic regulatory basis, which is supported by the pioneering study on the characterization of a peach evergrowing (EVG) mutant that loses dormancy in both apical shoot meristems and floral buds23. Genome analysis revealed six tandemly duplicated highly conserved dormancy-associated MADS-BOX genes (DAM1-6) located in the Wt EVG locus, and identified a large deletion removing the DAM1-4 and silencing the adjacent DAM5-6 within the mutant evg locus24, thus providing compelling evidence that loss of expression of six DAM genes leads to the EVG phenotype. Gene expression analyses showed that in peach and other Prunus species, DAM1, DAM2, and DAM4 are upregulated in apical leaf meristem during late summer and early fall, coincident with its growth cessation and bud formation, a stage of ecodormancy, while DAM5 and DAM6 are increased throughout fall, coincident with the transition from ecodormancy to endodormancy25. These data suggest that these DAMs differentially regulate the dormancy onset, which is further supported by a transgenic study where ectopic expression of DAM6 in poplar promoted growth cessation, bud set and a prolonged dormancy period26. In almond (Prunus dulcis), PdDAM6 showed a continuous decrease in transcript levels for both cultivars with different chilling requirements and flowering time during its dormancy release27.
Several lines of evidence also support that DAMs serve as direct targets of chilling temperatures. First, a major QTL trait responsible for chilling requirement was mapped to the peach EVG locus28. Second, DAM5 and DAM6 are downregulated during the chilling period or dormancy release11. Third, application of hydrogen cyanimide that promotes dormancy break in peach also downregulates DAM5 and DAM6 in dormant vegetative and floral buds29. Fourth, cultivars with a transposon insertion in both DAM5 and DAM6 require less chilling30. Thus, downregulation or genetic mutation of specific DAM genes is correlated to dormancy release or reduced chilling requirement. Given that DAMs are homologous to Arabidopsis short vegetative phase, a gene that codes for a transcription repressor that specifically targets, in parallel to flowering locus C (FLC), the flower and organ identify genes31,32,33, seasonal oscillation of DAMs could directly orchestrate the flower developmental course coupled with dormancy entry and release: Increased expression in late summer to fall slows down or arrests the floral developmental course (dormancy entry) but decreased expression during winter releases such arrest (dormancy release).
Considering variation of chilling requirement between vegetative and floral buds and male and female organs, how the cluster of six DAM genes in peach regulates bud- and organ-specific chilling requirement and dormancy onset and release remains unknown. In addition, whether all DAMs are similarly or differentially regulated in the same bud or tissues is not known. In Arabidopsis, chilling is known to directly target FLC, a transcriptional repressor, through induction of histone methylation, H3K27me334, via cold assisted intronic noncoding RNA (COLDAIR) that assists the deposition of H3K27me3 on the FLC chromatin35. Whether DAMs share similar or different epigenetic regulation with FLC remains to be studied. In this study, we set out to address these questions and understand how DAMs are genetically and epigenetically regulated by chilling and the ensuing warm temperature regimes.
Results
Assessment of chilling requirement in the evergrowing (EVG) mutant peach
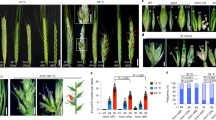
Although the peach EVG mutant loses dormancy onset in the apical shoot meristem and floral buds, its lateral leaf buds appear to retain dormancy and the chilling requirement23, raising a question of whether DAMs are exclusively involved in regulation of dormancy and chilling requirement in all buds. To address this, we assessed the chilling requirement and dormancy release of the lateral leaf and floral buds by directly placing the cut shoots from the evg trees under permissive growth conditions (~20 °C) without chilling treatment. In parallel, the cut shoots with fully dormant leaf buds and floral buds from Wt peach cultivar “John Boy” were included as a control. It is noted that the evg tree only formed the floral and lateral leaf buds but apical meristems still remained actively growing at the time when samples were collected. Therefore, this experiment was performed on wild-type and mutant shoots with or without the removal of the apical meristems. The floral and lateral leaf buds from the evg tree should, if completely free from dormancy constraints, continue the course of growth or development and progress into bud break instead of arrest. Figure 1a shows that all Wt buds (e.g., apical and lateral leaf and floral buds) remained arrested even after 40 days under the permissive condition (left panel) unless given prior treatment of 1000 h chilling (Fig. 1b–d). But both lateral leaf and floral buds from the evg tree continued to break, grow, and develop (right panel, Fig. 1a). Our work provides compelling evidence that the loss of DAMs or their expression in the evg trees abolishes dormancy and chilling requirement not only in the apical shoot meristems but also in the lateral leaf and floral buds as well.

a Confirmation of chilling-independent bud break in the peach evg mutant. The shoots attached with leaf and floral buds collected from the evg (right) and Wt (left) peach trees in the late fall were directly placed under normal growth condition (~ 20 °C) for 40 days without chilling treatment. LFB lateral leaf bud, FB floral buds. b–d In vitro chilling treatment of fully dormant Wt peach floral buds at 4 °C for 0 h (T1, b), 500 h (T2, c), and 1000 h (T3, d), and then transferred to the normal growth condition at greenhouse (~20 °C) for 14 days for assay of bud break. e The morphology of the chilled T1, T2, and T3 floral buds as well as the chilled T3 buds in greenhouse (~20 °C) for 3 days (D3) and 7 days (D7). f RNA-seq analysis of DAM expression plotted with RPKM (reads per kilobase per million mapped reads). Data are averaged from three biological replicates, with ±SD. g The percentage of total up- and downregulated genes from stage to stage. h Dissection and collection of petal (Pe), stamen (St), and carpel (Ca) tissues from dormant T1–T3 floral buds. i RNA-seq analysis of floral organ-specific expression. Data are averaged from three biological replicates, with ±SD. TPM transcripts per kilobase million mapped reads
Chilling differentially downregulated five of six DAMs in dormant floral buds with identification of DAM4 as the most abundantly expressed one
To understand how the six DAMs were regulated by chilling during dormancy release in floral buds, we analyzed their expression and regulation in wild-type cultivar “John Boy” dormant floral buds after treatment with chilling conditions (4 °C) for 0, 500, and 1000 h (T1, T2, and T3, respectively) before being transferred to a permissive growth condition (20 °C) to induce bud break (as shown in Fig. 1b–d). This in vitro assay of the chilling requirement has been developed and used for several decades36, enabling accurate assessment of the effect of chilling and the following warm temperatures on dormancy release or bud break without the complications of drought, freezing, and/or transient warm spells that often occur under the field conditions. Only fully chilled T3 buds (1000 chilling hours (CH)) flowered after being transferred to the permissive growth conditions of (20 °C) in the greenhouse (Fig. 1b–d). Morphologically, the chilled T3 buds underwent slight enlargement at 3 days (D3) post warm temperature treatment and developed full petals at 7 days (D7) (Fig. 1e), indicating that 1000 CH completely releases the floral buds from dormancy constraints. On the contrary, neither T1 nor T2 chilled buds showed apparent morphological changes and did not exhibit bud break (Fig. 1b, c, e). Accordingly, we collected floral tissue (absent bud scales) from the treated buds at the T1, T2, T3, D3, and D7 stages (Fig. 1e) for RNA-seq, strand-specific RNA-seq (ssRNA-seq), small RNA-seq (sRNA-seq), whole genome bisulfite sequencing (BS-seq), and ChIP-seq analyses described below.
First, we found that chilling significantly downregulated five (DAM1, 3, 4, 5, and 6) of the six DAMs (FDR < 0.05), while DAM2 expression was consistently low and near the limit of detection (Fig.1f). Interestingly, the five DAMs showed chilling stage-specific downregulation patterns. DAM5 and DAM6 progressively decreased from T1 to T3, while DAM1 and DAM3 sharply dropped from T1 to T2 and DAM4 precipitously declined from T2 to T3. The relative expression levels also varied greatly among DAMs. DAM4 was the highest of all DAMs at T2 (~512 RPKM) followed by DAM3 at T1 (~170 RPKM), and DAM5 and DAM6 at T1 (~30 RPKM), while the lowest level was reached by DAM1 at T1 (only ~7 RPKM). Thus, the expression of DAM4 was at least 3 times more abundant than DAM3, 17 times more abundant than DAM5 and DAM6, and 70 times more abundant than DAM1. Similarly, downregulation of DAM4 from 512 RPKM at T2 to about 30 RPKM at T3, represented about a 17-fold reduction, the most profound observed change compared to the relatively small amplitude of reduction of DAM1, 3, 5, and 6 transcript abundance. Following warm conditions (20 °C), DAM4, 5, and 6 were further downregulated, while DAM1 and 3 remained unchanged or slightly upregulated, suggesting that the warm treatment strongly reinforced the chilling-imposed repression on DAM4, 5, and 6. To rule out the possibility that repression of DAMs may result from ubiquitous, genome-wide transcriptional repression imposed by chilling stress, we analyzed genome-wide differentially expressed genes (DEGs) during the chilling period (Table S1). Over 60% of DEGs were upregulated by chilling from state to state (T2 vs T1, T3 vs T2, T3 vs T1, and D7 vs T3) (Fig. 1g), confirming that the downregulation of DAMs were biologically specific rather than result from global repression induced by chilling stress.
DAM3 and DAM4 were preferentially expressed in carpel
To understand whether the DAM expression was potentially flower organ specific, we isolated carpel, petal, and stamen from T1 to T3 flowers (Fig. 1h), respectively, and pooled them for transcriptome analysis. Consistent with whole flower data presented in Fig. 1i, DAM4 was the most abundantly expressed in three floral organs followed by DAM3, while DAM1, DAM5, and DAM6 exhibited the lowest expression (Fig. 1i). However, expression levels of both DAM3 and DAM4 were almost two or three times higher in carpel tissues than petal and stamen, respectively, (Fig. 1i). Interestingly, DAM1, 5, and 6, while expressed at lower levels, had relatively higher expression in carpels.
Differential response of sRNAs produced in the DAM regions to chilling and warm treatments
Since sRNAs are known to act as either transcriptional or post-transcriptional regulators, we next examined whether the chilling and warm temperatures also regulated sRNA production in DAMs. Figure 2a shows that sRNAs were produced from all six DAMs as well as the 10-kb intergenic region (ITGR) that separates DAM1 and 2 from DAM3, 4, 5, and 6 (Fig. S1). sRNAs remained little changed from T1 to T3 in the six DAMs and ITGR region but were substantially increased in all except DAM1 and ITGR from T3 to D7, indicating a warm-responsive induction. We then analyzed size (20–26 nt) of sRNAs produced in each DAM and their responses to the treatments. Figure 2b–f shows that 24-nt sRNA was the predominant species in all regions analyzed. Interestingly, the DAM3 region also produced an additional 21-nt sRNA species as a minor group induced from T1 to T2 (Fig. 2b, c) and further elevated at T3 (Fig. 2d) before sharply declining at D3 (Fig. 2e), which is opposite to chilling-induced downregulation of the cognate DAM3 (Fig. 1g). To locate where individual sRNAs were produced in DAMs, we mapped sRNA reads against the 65-kb DAM genomic region. sRNA production preferentially occurred in 34 putative sRNA-producing regions or loci (highlighted) that were classified based on the shared expression pattern (Fig. 2g). Seven of them were in DAM1 and DAM3, six in DAM2, five in DAM5 and DAM6, and four in DAM4, respectively. The Sr loci varied in size, ranging from 43 to 592-bp but most of them were shorter than 250-bp (Table S2). Intriguingly, only three Sr loci were located within the putative promoter (Sr14 in DAM3) or transcriptional terminus (Sr13 in DAM2 and Sr34 in DAM6). The remaining 31 resided within the transcribed regions. Of those 31 loci, 25 were located within introns and the remaining six resided either in the intron–exon junctions (Sr19 in intron 7–exon 8 and Sr20 in intron 8–exon 9 of DAM3, and Sr24 in intron 8–exon 9 of DAM4), or in the last two exons (Sr5, Sr6, and Sr7 in exon 9 of DAM1, and Sr28 in the exon 8 of DAM5). To verify that Sr loci were independently transcribed, we performed RNA gel blotting analysis and detected 24-nt sRNA production in the representative Sr10, 14, 29, and 33 loci, respectively, but neither of them shared the same expression pattern from T1 to D7 with each other or with their cognate DAMs (Fig. 2h), confirming an independent regulation.

a sRNA expression in six DAMs. Data are averaged from three biological replicates, with ±SD. b–f sRNA population (20–26 nt) and dynamic change in DAMs and the intergenic region (ITGR) at T1 (b), T2 (c), T3 (d), D3 (e), and D7 (f). g sRNA mapping and sRNA-producing regions (Srs)/loci in six DAM regions. The region with similar sRNA expression activity was grouped as a sRNA-producing region (Sr)/locus, and the 34Sr loci are highlighted, depicted, and marked. Red specifies antisense transcript reads and green specifies sense transcript reads. Annotated six DAMs and ITGR are depicted above. h RNA gel blotting. About 20 µg of enriched small RNA sample along with RNA marker was resolved on 16% of PAGE gel, blotted, probed, and reprobed with indicated p32-labeled oligos together with the labeled RNA marker. Sizes (21 and 25 nt) of sRNA markers are indicated at left and U6 serves as a control. i–p sRNA expression of individual Sr locus in DAM1 (i), DAM2 (j), DAM3 (k), DAM4 (l), DAM5 (m), and DAM6 (n), respectively. Data are averaged from three biological replicates, with ±SD. oSr loci upregulated by chilling. p 21-nt sRNA predominantly expressed at the Sr16 locus
Chilling preferentially induced 21-nt sRNA of Sr16 located at DAM3, while warm upregulated many Sr loci coding for 24-nt sRNAs in different DAMs
We examined responses of the Sr loci to the chilling and warm treatments. First, all Sr loci displayed distinct expression trajectories during thermal treatment (Fig. 2i–n). Of 34Sr loci, 21 responded to the warm temperature from T3 to D7, with 15 upregulated (Sr1, Sr3, Sr5, Sr10, Sr14, Sr15, Sr21, Sr23, Sr24, Sr25, Sr26, Sr27, Sr29, Sr30, and Sr33) and six downregulated (Sr6, Sr8, Sr9, Sr22, Sr32, and Sr34), while eight (Sr 2, 4, 7, 11, 12, 13, 17 and, 31) did not respond to the chilling or warm treatments, most of which were located within DAM1 and DAM2. Five Sr loci (Sr16, 18, 19, 20, and 28) responded to the chilling treatment: Sr16, 19, and 28 were upregulated and Sr18 and Sr20 downregulated. In particular, Sr16 was the most abundant and showed the strongest response to chilling compared to others (Fig. 2o). sRNA size analysis revealed that Sr16 was the only locus coding for a 21-nt sRNA (Fig. 2p). Hence, the chilling-induced 21-nt sRNA detected within DAM3 from T1 to T3 (Fig. 2b–d) was exclusively encoded by Sr16.
Chilling increased CHG and CHH methylation in DAM4 but the warm treatment differentially regulated methylation at DAMs in a sequence context-dependent manner
Given that 24-nt sRNAs guide DNA methylation through RNA-dependent DNA methylation37, we performed whole genome BS-seq to ascertain whether chilling and warm temperatures regulate the methylation of cytosines at DAMs. Figure 3a shows that CG, CHG, and CHH (where H = A, T, or C) sequence contexts at DAMs were overall hypermethylated but the methylation trajectories responded differently to the chilling and warm temperatures. The CG methylation in all regions analyzed remained relatively constant during the chilling period (T1 to T3) but declined after shifting to the warm temperature (T3 to D7). CHH methylation changed little during chilling in all regions except DAM4, in which an increase in CHH methylation was observed. However, the warm temperature increased CHH methylation overall across all regions except DAM1 and this increase was particularly pronounced in DAM4 and DAM5. The increase was also correlated with an increase of sRNA production in these same DAMs (Fig. 3a). Evidently, the warm temperature appeared to oppositely regulate CG and CHH methylation. The effect of the chilling temperature on CHG methylation was diverse. It appeared to increase the CHG methylation in DAM4 but decrease in DAM1 and DAM3, and change little in DAM2, 5, and 6. It became apparent that DAM4 was only gene that was up-methylated by chilling at CHH and CHG contexts and by warm at CHH context.

a CG, CHG, and CHH methylation in six DAMs, ITGR, and peach genome. Methylation rate (%) was indicated on the left y-axis and sRNA abundance (orange) expressed by Log2 RPKM on the right y-axis. The treatment time point T1, T2, T3 and D7, and DAM genes are indicated at the bottom. b Cluster of methylated regions (Mrs). A total of 44 Methylated regions (Mrs) are marked in the bottom, while the corresponding Sr regions highlighted in yellow and denoted on top. Number of methylated regions starts from DAM1 to DAM6 for the sake of consistence with DAM position. Green—CG methylation. Purple—CHG methylation. Red—CHH methylation. The Mrs independent of siRNAs are named as downstream or upstream of adjacent Mrs (e.g. Mr2u, Mr17u, Mr18u, Mr18d, etc.)
Overlap of the methylated region (Mr) with the Sr region
We then investigated whether DNA methylation overlapped with the Sr loci or closely associated with sRNA production. Figure 3b shows that methylation was not randomly distributed in DAMs but rather clustered in approximately 44 regions, dubbed Mrs. The majority of the Mr loci overlapped with the Sr loci except Sr1, 19, and 28 where there was no methylation detected. These results are indicative of widespread occurrence of RdDM in DAMs. The Mr regions were typically larger than their corresponding Sr loci (Table S2), consistent with methylation spreading to flanking regions38. Ten additional Mr regions (e.g., Mr1u, Mr2u, Mr17u, Mr18u, Mr18d, Mr22u, Mr23d, Mr24d, Mr26u, and Mr32d) located either upstream or downstream of the Sr-overlapped Mr loci, shared no overlap with any Sr region, signifying the occurrence of siRNA-independent methylation, a phenomenon frequently observed in plant genomes. Taken together, methylation regulation at the Mr regions in different sequence contexts under the chilling and warm conditions was diverse and complex but the overall correlation between the CHH methylation level and sRNA abundance under the warm condition was apparent in many Mrs (Fig. S2).
Chilling and warm differentially induced H3K27me3 and its spreading
Earlier studies showed that chilling-induced H3K27me3 in some DAMs during dormancy release39. We performed ChIP-seq to understand how chilling and warm temperatures regulated H3K27me3 in all six DAMs. The ChIP-seq reads were enriched during the chilling period in DAM1, 2, 5, and 6 and some parts of the ITGR region but the enriched patterns and locations differed among them (Fig. 4a). Small, localized enrichment occurred in either intron or exon regions of DAM1, 2, 5, and a few regions of ITGR but were gene-wide in the entire DAM6 transcribed region. Furthermore, the timeframe of the occurrence of the read enrichment varied as the enriched peak appeared only at T2 onward in DAM1 and 5 but at T1 onward for DAM2, 6 and ITGR, indicating that the read enrichment in DAM1 and 5 is chilling-dependent (Fig. 4a). At the warm temperature from T3 to D7, the read enrichment patterns in DAM2 and DAM6 along with ITGR remained little changed but those within DAM1 and 5 underwent evident changes, with localized peaks becoming large to cover the entire transcribed regions, and was particularly robust in DAM5 (bottom panels, Fig. 4a). Interestingly, DAM4, despite a lack of visible enrichment peak during the chilling period, also showed a major peak near the transcription start region at D3 and spread toward the 3′ end by D7 (Fig. 4a), indicating warm induced and facilitated the spreading of H3K27me across the DAM4 region. Quantitative read enrichment analysis further confirmed that the chilling and warm conditions significantly induced and enhanced H3K27me3 in DAM1 and DAM5, while warm did in the DAM4 region (Fig. 4b). The warm temperature also enhanced H3K27me3 at DAM3 and DAM6 even though it was not clearly as discernable in the read map (Fig. 4a).

a Genome browser tracks showing H3K27me3 ChIP-seq reads are mapped to the 65-kb DAM region. Five ChIP-seq tracks from T1 to D7, and their no-antibody control tracks (Ctr) are shown. The peaks in DAMs are marked by blue boxes. b Quantitative analysis of the ChIP-seq read counts in DAMs and ITGR. Data are averaged from two biological replicates, with ±SD
Chilling induced a noncoding RNA (ncRNAs) in the DAM4 region
Strand-specific RNA-seq was carried out to search for ncRNAs corresponding to DAMs. As shown in Fig. 5a, abundant sense reads specifically mapped to the second introns of DAM3-5, which roughly corresponded to 366-, 354-, and 235-nt regions, respectively, (Fig. 5b). Accordingly, we named these as putative D3ncRNA, D4ncRNA, and D5ncRNA, respectively. The three ncRNAs were situated at similar locations in the second intron (Fig. 5a, b) and shared ~70% of sequence identity but were differentially regulated by chilling (Fig. 5c). D3ncRNA and D5ncRNA remained either unchanged or downregulated, while D4ncRNA was drastically upregulated from T1 to T2 and reached maximal expression level at T3 (Fig. 5c). We then compared the expression of D4ncRNA with its cognate DAM4 and revealed a strong inverse correlation during the chilling treatment. At the warm temperature, expression of both DAM4 and D4ncRNA quickly dropped, suggesting that a common mechanism may operate to repress both at these stages. Given that chilling-inducible COLDAIR located in the largest intron of Arabidopsis FLC is transcribed by its own 109-bp promoter35, we examined epigenetic changes in the putative promoter region located upstream of the D4ncRNA region. Coincidently, a 588-bp Mr21/Sr21 locus (described above) was located 25 bp upstream of the D4ncRNA-coding region and would be anticipated to overlap with the D4ncRNA promoter region (Fig. 5b). The Mr21/Sr21 locus region also overlapped with a major peak of H3K27me3 in DAM4 at the warm temperature (Fig. 4a). Accordingly, we focused on Mr21/Sr21 and found that 24-nt sRNA expression, CHH and CHG methylation and H3K27me3 all remained at a low or a moderately low level during the chilling but were rapidly upregulated from T3 to D7 (Figs. 4a, b and 5e). Apparently, the increased sRNA, DNA methylation and H3K27me3 in the putative promoter or Mr21/Sr21, collectively correlated with a strong repression of D4ncRNA at the warm temperature.

a Mapping of strand-specific RNA-seq reads against six DAMs. The sense read relevant to the DAM transcription direction is denoted in green and antisense in red. The detailed information about DAM organization and location is illustrated in Fig. S1 legend. Note that for the sake of clarity, the reads mapped to exons were removed. b Schematic diagram of the position and size of ncRNAs and relevant Mr/Sr locus. c Expression of three ncRNAs. RPKM—reads per kb per million mapped reads. d plot D4ncRNA expression from strand-specific RNA-seq data (SS-RNA-seq) with DAM4 expression from regular RNA-seq data. e DNA methylation and sRNA expression in Mr/Sr21 locus
Discussion
The coupling of flower development with dormancy onset and exit cycles in response to seasonal temperatures represents a key adaptive strategy that plants evolved to cope with temperature stress in order to ensure successful reproduction. Here, we show that the chilling and successive warm temperatures regulate diverse epigenetic events that may synergically enable DAMs rapidly downregulated under the chilling condition and irreversibly repressed during growth seasons, allowing the flower developmental pace successfully proceeded and flower buds breaking at a proper time or season.
Identification of DAM4 as a key regulator in the floral buds
Earlier studies showed upregulation of DAM1, 2, and 4 during the growth cessation of the shoot apical meristem and bud initiation, a stage of ecodormancy, and upregulation of DAM5 and 6 during transition from ecodormancy to endodormancy during fall, and downregulation of DAM5 and DAM6 during winter period25. Collectively these findings suggest that these DAMs differentially regulate dormancy onset, development and release in apical leaf buds. In the floral and lateral buds, DAM5 and DAM6 were also shown to be downregulated by chilling during dormancy release11,26,29, suggesting that DAM5 and DAM6 are involved in chilling requirement and dormancy release in those buds as well. Here, our analyses revealed complex regulation of DAMs in peach floral buds and showed that five of the six DAMs were downregulated by chilling with distinct patterns (Fig. 1f). Further, we also identified DAM4 rather than DAM5 or DAM6 as the most abundantly expressed DAM gene, with at least 3–17 times higher expression than the other four DAMs (Fig. 1f). DAM4 was expressed preferentially in carpel (Fig. 1i) and was mainly downregulated at the late stage of the chilling period (Fig. 1f), which is estimated to slightly precede the formation of the ovule in the carpel, a key stage of female organ development14,15,16,40. Given the overall correlation of DAM4 expression with dormancy exit and the corresponding unique and pronounced epigenetic events documented within DAM4, we propose DAM4 as a potential key dormancy regulator in floral buds. DAM4 exhibited 17-fold reduction of expression by chilling and remained at a considerable level (~30 RPKM) equivalent to the highest expression exhibited (~30 RPKM) by DAM5 or DAM6 at the T1 stage (Fig. 1f). At the warm temperature, DAM4 continuously declined and reached the lowest level at D3 onward, indicating that the warm further downregulates DAM4. A predominant role of DAM4 in floral bud dormancy and bud break is also supported by recent QTL mapping analysis28, which showed that Pchgms40, a marker located in DAM4, is more tightly linked to the traits of chilling and heat requirement and bud break compared with Pchgms12 located at DAM6 or Pchgms41 located at DAM5, respectively. Taken together, DAM4 apparently acts as a key regulator and source of trait variability for the chilling and heat requirement and bud break in peach floral buds.
It is noted that DAM3 also shows carpel-preferential expression and to a lesser degree DAM1, 5, and 6, indicating that these DAMs likely play an important though lesser role in modulating dormancy and bud break phenotypes as well. The shared carpel-preferential expression among five DAMs would suggest that they may be similarly regulated due to their conserved sequence and duplicated nature. Given a short intergenic region (~500–720 bp) exists between adjacent DAMs (Fig. S1), the key regulatory elements or enhancers should, like many of them in MADS BOX genes41,42, be situated in one of the introns. Alternatively, six DAMs could be co-regulated by a single enhancer that activates or represses DAMs at a distance. This enhancer could, if potentially, reside in the DAM4 region because deletion of DAM1-4 in the EVG mutant abolishes the DAM5 and DAM6 expression24. As might be expected if this were the case, the DAMs flanking DAM4 have reduced expression proportional to their distance to DAM4 (Figs. 1f, 2g, and S1).
Chilling drives distinct epigenetic interactions that define unique regulatory trajectories for each DAM
In Arabidopsis, chilling induces a ncRNA and H3K27me3 to silence FLC34,35. Another recent study on almond provides information on coding regions linked to early and late flowering methylation markers. It is also found that the methylation state of ten gene-coding sequences is linked to the dormancy release process43. In peach, chilling induces similar epigenetic responses, which vary among the six DAMs, with H3K27me3 induced in DAM1, 5, and 6, and Sr16 sRNA in DAM3 and D4ncRNA and CHG and CHH methylation in DAM4, respectively. None of these DAMs shares the same epigenetic regulation with each other or with Arabidopsis FLC, indicating DAM-specific epigenetic regulation. However, all DAMs are ubiquitously hypermethylated (Fig. 3a, b), and such hypermethylation may add an additional layer of repression for each DAM. Hence, interactions between hypermethylation and other epigenetic elements should contribute to variation of expression abundance and pattern among DAMs.
Previous work showed that H3K27me3 in DAM1, 4, 5, and 6 in floral buds is upregulated during dormancy release under field conditions39, but the absence of clearly defined chilling and warm periods and dormancy state makes it hard to discriminate the role of chilling from the successive warm temperature. Our work shows that chilling and warming function distinctly: Chilling induces the localized H3K27me3 in DAM1 and 5, while the warm enhances and spreads it gene-wide. In DAM4, only warm but not chilling induces H3K27me3. However, H3K27me3 in DAM6 occurs before chilling treatment and remains almost unchanged from the beginning of chilling to end of the warm period (Fig. 4a, b), apparently contradicting with the earlier finding that H3K27me3 in DAM6 is upregulated during dormancy release39,44. Hence, the chilling and warming effect on H3H27me3 varies among DAMs and possibly differently among peach cultivars. Interestingly, the presence of H3K27me3 is, regardless of abundance and stages, associated with lower expression (e.g. DAM1, 5, and 6), while the absence of it is correlated with higher expression (e.g. DAM3 and 4) during chilling period (Fig. 1f). This could be interpreted to imply that H3K27me3 presents an overall repressive effect on DAMs or that the lower expressed DAMs may be readily prone to H3K27me3.
Chilling-induced COLDAIR in the Arabidopsis FLC is involved in recruitment of a protein complex that deposits H3K27me335, but lack of detectable H3K27me3 in DAM4 during the chilling period indicates that the chilling-induced D4ncRNA functions differently in DAM4. ncRNAs have been shown to regulate H3K9 methylation45,46, histone deacylation,47 and recruitment of protein or transcription factors48,49. D4ncRNA may be involved in one of these regulatory events to downregulate DAM4. The role of the chilling-induced 21-nt sRNA coded by Sr16 in DAM3 remains mysterious and it may directly or indirectly repress the DAM3 expression transcriptionally or post-transcriptionally.
Warm treatment reinforces the chilling-imposed epigenetic repression on DAMs
The fact that warming reinforces the chilling-induced, localized H3K27me3 in DAMs (Fig. 4a, b) is consistent with the effect of warming on enhancement of the chilling-induced H3K27me3 in FLC in the vernalized Arabidopsis plants50,51. Such enhancement is believed to stabilize and reinforce repression of FLC over the growing season52. Expectedly, the warm-induced upregulation of H3K27me3 in peach floral buds also reinforces the repression of the DAM genes. In addition, we also show that warming corresponds with upregulation of the CHH methylation, which is particularly apparent in DAM4, 5, and 6 (Fig. 3a–d). The increased methylation is correlated with 24-nt sRNA expression, indicating that warming may activate RdDM in DAMs. The concurrence of H3K27me3 with CHH methylation in the same DAMs should synergistically impose a stronger repression than either alone, which is supported by continuous downregulation of DAM4 and steadily repression of DAM5 and 6 during the warm period (Fig. 1f) because all three DAMs are subjected to stronger H3K27me3 and CHH methylation compared with DAM1, 2 and 3 (Figs. 3a and 4a, b).
The warm-induced and reinforced epigenetic repression on DAMs is biologically important
In the winter Arabidopsis ecotype, a few weeks of a warm period (~20 °C) immediately following vernalization/chilling treatment is critical for establishing and stabilizing vernalization/chilling effects50. The vernalized/chilled plants lose their commitment to flowering when immediately placed at 30 °C but remains committed to flowering when placed at 20 °C for 2 weeks before being transferred to 30 °C. Thus, the warm period at 20 °C following vernalization/chilling is critical for stabilizing or reinforcing “the vernalized state.” A similar phenomenon is also observed in peach flower buds21. The fully chilled flower buds usually develop normally at the warm temperature at or below 20 °C but abnormally at or above 25 °C. The higher temperature often causes the arrest of reproductive organs especially the embryo sac and results in poor fruit set21. Molecular analysis revealed that the chilling-induced H3K27me3 at the FLC chromatin in the vernalized plants is strongly enhanced at the ensuing warm treatment (20 °C or below) but substantially reduced at 30 °C unless prior exposure to 20 °C for 2 weeks50,51, which supports the role of warming (20 °C) in epigenetic reinforcement of the chilling-induced vernalization state. Hence, the warm-enhanced H3K27me3 and CHH methylation in the peach floral buds should similarly impose a stronger and stable repression on DAMs, but higher temperatures (≥25 °C) could abrogate such repression, leading to ectopic expression of DAMs in carpels and compromising the formation or development of normal embryo sac and other tissues21. To this end, the rate and efficiency of epigenetic response to the warm temperature to achieve a strong and irreversible repression on DAMs could vary among different species or cultivars, resulting in the different warm period or total heat sum required for bud break or flowering.
Materials and methods
Chilling treatment of dormant peach floral buds
Shoots from peach cultivar “John Boy” (Wt) and EVG were collected from trees grown in USDA-ARS orchard located at Kearneysville, WV, at the end of October when full dormant state in the buds are developed. The collected shoots were directly placed at 20 °C for assay of bud break for up to 8 weeks, or in containers filled with 1/5 water and chilled at 4 °C in the growth chamber in dark for 0, 500, and 1000 h (CH). The flower tissues were collected by dissecting and removing bud scales that enclose the buds, at three different stages: 0 (T1), 500 CH (T2), and 1000 CH (T3). After chilling treatment, the shoots were placed in the greenhouse (~20 °C) for assay of bud break, and the flower buds from shoots kept for 3 (D3) and 7 (D7) days in the greenhouse were collected. Three replicates were conducted for each time point of sample collection. The same tissues were used for isolation of RNA and DNA for RNA-seq, BS-seq, sRNA-seq, and ChIP-seq analyses described below. The petal, carpel, and stamen tissues were also dissected and isolated from T1, T2, and T3 buds and pooled the same tissue together for analysis of floral organ-specific regulation of DAMs.
RNA-seq and strand-specific RNA-seq data analyses
Total RNA samples with three biological replicates were isolated from the flower tissues or organ tissues. About 200 mg of the floral tissues were grounded in liquid nitrogen and extracted using TriReagent (Sigma, St Louis, MO, USA) followed by two rounds of phenol–chloroform extraction (50:50). About 5 μg of each sample was submitted to BGI Americas Cooperation (Cambridge, MA, USA) for RNA-seq and ssRNA-seq using Illumina Hi-Seq. RNA-seq reads were first processed by removing the 3′ adapter sequence, filtering out rRNA and tRNA sequences via CLC Genomic Workbench V.5, 20 (Qiagen, Hilden, Germany). Floral organ-specific expression and analysis of ncRNAs and expression were conducted by directly mapping the resulting filtered reads to the 65-kb region accurately annotated with the six DAMs based on Peach genome 1.053 (also see Fig. S1). The read counts in each DAMs or ncRNA region were normalized to reads per transcript per million mapped reads or reads per kilobase of exon model per million mapped reads (RPKM), respectively. Differential gene and transcript expression analysis were conducted according to instruction provided by CLC Bio (Qiagen, Hilden, Germany), and Raw P values of multiple tests were corrected using FDR54.
ChIP-seq and analysis of H3K27me3 in DAMs
About 200–300 mg of the harvested peach flower bud tissues were ground to fine power under liquid nitrogen, and the nuclei isolation and ChIP-seq were performed as previously described55. The isolated chromatins were digested by micrococcal nuclease (NEB, Ipswich, MA, USA). Half of the nucleosomes was directly used for library construction and sequenced as a negative control, while the other half was subjected to immunoprecipitation using antibody against H3K27me3 (Millipore 07-449). About 0.5 to 1 µg of the recovered immunoprecipitated DNA samples were submitted to the core facility of Cornell Weill Medical College for library construction and sequencing on HiSeq2000 platform. Two replicates for each sample were performed. The ChIP-seq reads were mapped using bowtie and peak calling was performed using MACS2 and CLC platform (Qiagen, Hilden, Germany). The reads per kilobase per million mapped reads (RPKM) were calculated and statistically analyzed as presented in Fig. 4b.
RNA Gel blotting
RNA blot analysis was carried out as described previously by Zhu et al56. Briefly, total RNA was extracted from peach floral buds using TriReagent (Sigma, St Louis, MO, USA) followed by two rounds of phenol–chloroform extraction (50:50). sRNAs were further enriched using the mirVana miRNA isolation kit (AM1560, Thermo Fisher Scientific), and about 25 µg of the enriched sRNA was blotted on to the nylon membrane filters, which were probed and re-probed with 32P-labeled Sr-specific probes together with the sRNA Marker Probe labeled with γ32P-ATP using T4 polynucleotide kinase (NEB, Beverly, MA, USA).
Small RNA sequencing and read alignment
Small RNAs were isolated and enriched from total RNA as described above, and ligated to a 5′ RNA adapter and a 3′ RNA adapter, as described previously57. The ligation product was RT-PCR amplified and gel purified before sequencing on Illumina HiSeq 2000 platform. Three biological replicates were sequenced. Adapter sequences were first removed from raw sRNA reads. The resulting sRNA sequences were further processed to remove those containing low-complexity and t/rRNA sequences, and having lengths <15 bp or >29 bp. The remaining high-quality sRNA reads were aligned to the peach genome 1.0 and the 65-kb DAM sequence with perfect matches and reads with multiple alignments in the genome were excluded from further analysis. Raw read counts for each sRNA were normalized to RPKM and statistical analysis of changes of all sRNAs along DAM region (Sr) during temperature-dependent dormancy release and flowering was performed, using CLC Genomic Workbench V.5 (Qiagen, Hilden, Germany).
Whole genome bisulfite sequencing and data processing
DNA samples with three replicates were isolated from flower tissues using the DNeasy® Plant Mini Kit (Qiagen, Hilden, Germany) and submitted to BGI Americas Cooperation (Cambridge, MA, USA) for whole genome BS-Seq, with about 30X genome coverage. The error conversion rates of the BS sequences were below 0.005% for unmodified cytosines, comparable to previous data58. To align the BS-seq reads to peach genome, cytosine bases in the reads were first replaced with thymines. The converted reads were then aligned to the computationally converted strands of the peach genome 1.0 (one with C to T and the other with G to A), respectively, using the Bowtie algorithm allowing up to two mismatches59. Alignments from both strands were combined, and for each read only the optimal alignments were kept. Multialigned reads were not included in the analysis. The read sequences in the alignments were then replaced with the original, nonconverted sequences59. Finally, methylation level of each cytosine was calculated genome-wide and the methylation profiling (in CG, CHG, and CHH contexts) was presented along DAM region, on the basis of alignments.
Data availability
All the raw data from this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/493230) under accession number PRJNA493230.
References
Arora, R., Rowland, L. J. & Tanino, K. Induction and release of bud dormancy in woody perennials: a science comes of age. HortScience38, 911–921 (2003).
Liu, Z., Zhu, H. & Abbott, A. in Advances in Plant Dormancy (ed. Anderson, J. V.) 75–105 (Springer, 2015).
Horvath, D. P., Anderson, J. V., Chao, W. S. & Foley, M. E. Knowing when to grow: signals regulating bud dormancy. Trends Plant Sci.8, 534–540 (2003).
Rohde, A. & Bhalerao, R. P. Plant dormancy in the perennial context. Trends Plant Sci.12, 217–223 (2007).
Lang, G. A., Early, J. D., Martin, G. C. & Darnell, R. L. Endo-, para-, and ecodormancy: physiological terminology and classification for dormancy research. HortScience22, 371–377 (1987).
Hillman, J. R. in Advanced Plant Physiology (ed. Wilkins, M. B.) 127–148 (Pitmann, 1984).
Erez, A., Couvillon, A. & Hendershott, C. H. Quantitative chilling enhancement and negation in peach buds by high temperatures in a daily cycle. J. Am. Soc. Hortic. Sci.104, 536–540 (1979).
Atkinson, C. J., Brennan, R. M. & Jones, H. G. Declining chilling and its impact on temperate perennial crops. Environ. Exp. Bot.91, 48–62 (2013).
Zhang, Y. X. et al. Differential expression proteins associated with bud dormancy release during chilling treatment of tree peony (Paeonia suffruticosa). Plant Biol.17, 114–122 (2015).
Campbell, M. A., Suttle, J. C. & Sell, T. W. Changes in cell cycle status and expression of p34cdc2 kinase during potato tuber meristem dormancy. Physiol. Plant.98, 743–752 (1996).
Yamane, H., Ooka, T., Jotatsu, H., Sasaki, R. & Tao, R. Expression analysis of PpDAM5 and PpDAM6 during flower bud development in peach. Sci. Hortic.129, 844–848 (2011).
Julian, C., Rodrigo, J. & Herrero, M. Stamen development and winter dormancy in apricot (Prunus armeniaca). Ann. Bot.108, 617–625 (2011).
Reinoso, H., Luna, V., Daurfa, C., Pharis, R. & Bottini, R. Dormancy in peach (Prunus persica L.) flower buds. VI. Effects of gibberellins and an acylcyclohexanedione (trinexapac-ethyl) on bud morphogenesis in field experiments with orchard trees and on cuttings. Can. J. Bot.80, 664–674 (2002).
Luna, V. et al. Dormancy in peach (Prunus persica L.) flower buds: I. Floral morphogenesis and endogenous gibberellins at the end of the dormancy period. Plant Physiol.93, 20–25 (1990).
Luna, V., Reinoso, H., Lorenzo, E., Bottini, R. & Abdala, G. Dormancy in peach (Prunus persica L.) flower buds. II. Comparative morphology and phenology in floral and vegetative buds, and the effect of chilling and gibberellin A3. Trees5, 244–246 (1991).
Luna, V., Soriano, M. D., Bottini, R., Sheng, C. X. & Pharis, R. P. Dormancy in peach (Prunus persica L.) flower buds. III. Levels of endogenous gibberellins, abscisic acid, indole-3- acetic acid, and naringenin during dormancy of peach flower buds. Acta Hortic.329, 265–267 (1993).
Ashcroft, G. L., Richardson, E. A. & Seeley, S. D. A statistical method of determining chill unit and growing degree hour requirements for deciduous fruit trees. HortScience12, 347–348 (1977).
Couvillon, G. A. & Erez, A. Effect of level and duration of high-temperatures on rest in the peach. J. Am. Soc. Hortic. Sci.110, 579–581 (1985a).
Eisensmith, S. P., Jones, A. L. & Flore, J. A. Predicting leaf emergence of ‘Montmorency’ sour cherry from degree-day accumulations. J. Am. Soc. Hortic. Sci.105, 75–78 (1980).
Cannell, M. G. R. in Manipulating of Fruiting (ed. Wright, C. J.) Ch. 99–113 (Butterworths, 1989).
Kozai, N. et al. Adverse effects of high temperature on the development of reproductive organs in ‘Hakuho’ peach trees. J. Hortic. Sci. Biotech.79, 533–537 (2004).
Nava, G. A. et al. Effect of high temperatures in the pre-blooming and blooming periods on ovule formation, pollen grains and yield of ‘Granda’ peach. Sci. Hortic.122, 37–44 (2009).
Rodriguez, A., Sherman, W. B., Scorza, R., Wisniewski, M. & Okie, W. R. Evergreen peach, its inheritance and dormant behavior. J. Am. Soc. Hortic. Sci.119, 789–792 (1994).
Bielenberg, D. G. et al. Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet. Genomes4, 495–507 (2008).
Li, Z., Reighard, G. L., Abbott, A. G. & Bielenberg, D. G. Dormancy-associated MADS genes from the EVG locus of peach [Prunus persica (L.) Batsch] have distinct seasonal and photoperiodic expression patterns. J. Exp. Bot.60, 3521–3530 (2009).
Sasaki, R. et al. Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant Physiol.157, 485–497 (2011).
Prudencio, A. S., Dicenta, F. & Martínez-Gómez, P. Monitoring dormancy transition in almond [Prunus dulcis (Miller) Webb] during cold and warm Mediterranean seasons through the analysis of a DAM (Dormancy-Associated MADS-Box). gene. Horticulturae4, 41 (2018).
Fan, S. et al. Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica). N. Phytol.185, 917–930 (2010).
Yamane, H. et al. Expressional regulation of PpDAM5 and PpDAM6, peach (Prunus persica) dormancy-associated MADS-box genes, by low temperature and dormancy-breaking reagent treatment. J. Exp. Bot.62, 3481–3488 (2011).
Yamane, H. et al. Comparative analyses of dormancy-associated MADS-box Genes, PpDAM5 and PpDAM6, in low- and high-chill peaches (Prunus persica L.). J. Jpn. Soc. Hortic. Sci.80, 276–283 (2011).
Jang, S., Torti, S. & Coupland, G. Genetic and spatial interactions between FT, TSF and SVP during the early stages of floral induction in Arabidopsis. Plant J.60, 614–625 (2009).
Lee, J. H. et al. Regulation of temperature-responsive flowering by MADS-box transcription factor repressors. Science342, 628–632 (2013).
Liu, Y., Koornneef, M. & Soppe, W. J. The absence of histone H2B monoubiquitination in the Arabidopsis hub1 (rdo4) mutant reveals a role for chromatin remodeling in seed dormancy. Plant Cell19, 433–444 (2007).
Bastow, R. et al. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature427, 164–167 (2004).
Heo, J. B. & Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science331, 76–79 (2011).
Couvillon, G. A. & Erez, A. Influence of prolonged exposure to chilling temperatures on bud break and heat requirement for bloom of several fruit species. J. Am. Soc. Hortic. Sci.110, 47–50 (1985b).
Zhang, H., Lang, Z. & Zhu, J. K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol.19, 489–506 (2018).
Tang, K., Lang, Z., Zhang, H. & Zhu, J. K. The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat. Plants2, 16169 (2016).
de la Fuente, L., Conesa, A., Lloret, A., Badenes, M. L. & Rios, G. Genome-wide changes in histone H3 lysine 27 trimethylation associated with bud dormancy release in peach. Tree Genet. Genomes11, 45 (2015).
Wang, L. et al. Impact of chilling accumulation and hydrogen cyanamide on floral organ development of sweet cherry in a warm region. J. Integr. Agric.11, 2529–2538 (2016).
Oliver, S. N., Finnegan, E. J., Dennis, E. S., Peacock, W. J. & Trevaskis, B. Vernalization-induced flowering in cereals is associated with changes in histone methylation at the VERNALIZATION1 gene. Proc. Natl Acad. Sci. USA106, 8386–8391 (2009).
Sieburth, L. E. & Meyerowitz, E. M. Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically. Plant Cell9, 355–365 (1997).
Prudencio, A. S. et al. DNA methylation analysis of dormancy release in almond (Prunus dulcis) flower buds using epi-genotyping by sequencing. Int. J. Mol. Sci.19, 3542 (2018).
Leida, C., Conesa, A., Llacer, G., Badenes, M. L. & Rios, G. Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. N. Phytol.193, 67–80 (2012).
Pandey, R. R. et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell32, 232–246 (2008).
Wagschal, A. et al. G9a histonemethyltransferase contributes to imprinting in themouse placenta. Mol. Cell Biol.28, 1104–1113 (2008).
McHugh, C. A. et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature521, 232–236 (2015).
Hung, T. et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet.43, 621–629 (2011).
Ng, S. Y., Bogu, G. K., Soh, B. S. & Stanton, L. W. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell51, 349–359 (2013).
Bouche, F., Detry, N. & Perilleux, C. Heat can erase epigenetic marks of vernalization in Arabidopsis. Plant Signal. Behav.10, e990799 (2015).
Angel, A., Song, J., Dean, C. & Howard, M. A Polycomb-based switch underlying quantitative epigenetic memory. Nature476, 105–108 (2011).
Sheldon, C. C. et al. Resetting of FLOWERING LOCUS C expression after epigenetic repression by vernalization. Proc. Natl Acad. Sci. USA105, 2214–2219 (2008).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet.45, 487–494 (2013).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc.57, 289–300 (1995).
Lv, P. et al. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat. Plants4, 784–791 (2018).
Zhu, H. et al. Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol.12, 149 (2012).
Lu, C., Meyers, B. C. & Green, P. J. Construction of small RNA cDNA libraries for deep sequencing. Methods43, 110–117 (2007).
Stroud, H., Greenberg, M. V., Feng, S., Bernatavichute, Y. V. & Jacobsen, S. E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell152, 352–364 (2013).
Guo, W. et al. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics14, 774 (2013).
Acknowledgements
We express our thanks to Mr. Dennis Bennett for performing RNA gel blotting and staffs at the Appalachian Fruit Research Station for providing peach materials used in this work. This study was partially funded by the ARS-IN-House fund, USDA-NIFA grant (3200000379-16-182), the National Natural Science Foundation of China (31772371) and AoE grant (AoE/M-403/16).
Author information
Authors and Affiliations
Contributions
Z.L. and H.Z. conceived and designed experiments. H.Z., P-.Y.C., S.Z., Y.Y., G-.Y.Z., and Z.L. conducted experiments. H.Z., P-.Y.C., S.Z., C.D., A.C., Y-.Q.A., S.v.K., A.A., and Z.L. performed data analyses and data interpretation. Z.L. wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, H., Chen, PY., Zhong, S. et al. Thermal-responsive genetic and epigenetic regulation of DAM cluster controlling dormancy and chilling requirement in peach floral buds. Hortic Res 7, 114 (2020). https://doi.org/10.1038/s41438-020-0336-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0336-y
This article is cited by
-
A high-quality assembled genome of a representative peach landrace, ‘Feichenghongli’, and analysis of distinct late florescence and narrow leaf traits
BMC Plant Biology (2023)
-
Evolutionary origin and functional specialization of Dormancy-Associated MADS box (DAM) proteins in perennial crops
BMC Plant Biology (2022)
-
Functional characterisation of five SVP genes in grape bud dormancy and flowering
Plant Growth Regulation (2022)
-
Bud endodormancy in deciduous fruit trees: advances and prospects
Horticulture Research (2021)
-
Regulatory circuits involving bud dormancy factor PpeDAM6
Horticulture Research (2021)

{kind=link}
{kind=link}