
Abstract
Juglandaceae species are plants of great economic value and have been cultivated, domesticated, and utilized by human society for a long time. Their edible, nutrient-rich nuts and tough, durable wood have attracted the attention of botanists and breeders. With the advent of the genomics era, genome sequencing of the Juglandaceae family has been greatly accelerated, and a large amount of data has been generated. In this paper, we introduce the Portal of Juglandaceae (PJU), a tool to bring all these data together. The PJU contains genomes, gene-coding sequences, protein sequences, various types of annotation information, expression data, and miRNA data, which are configured with BLAST, JBrowse, and our self-developed synteny analysis tool. The PJU has a user-friendly and straightforward interface that performs a variety of query tasks with a few simple operations. In the future, we hope that the PJU will serve as a hub for the study of the Juglandaceae family.
Similar content being viewed by others

Introduction
The Juglandaceae family, with eight genera and more than 60 species, has a wide distribution in North America, Europe, and Asia1. Throughout the history of human society, Juglandaceae species have been cultivated, bred, and harvested2. The two most well-known genera of Juglandaceae are Carya and Juglans. Most species in these two genera not only have edible, delicious, and highly nutritious nuts but also produce durable and decay-resistant wood3. For example, the timber of J. nigra (black walnut) is highly recognized as the highest grade hardwood in the western world4. The wood is characterized by its beautiful grain, dark color, high stability, and moderate density4.
Another highly valued Juglandaceae family commodity is nuts. The common walnut (J. regia), pecans (C. illinoinensis), and Chinese hickory (C. cathayensis) are the most consumed commercial nuts from the Juglandaceae family2. Compared to other commercial nuts, pecans and Chinese hickory contain high contents of dietary fiber, minerals, and vitamins5. The properties of a variety of antioxidants in these nuts (e.g., β-carotene) make them sought after in the global nut market6,7.
Although the Juglandaceae family occupies a very significant position in the economic landscape, there are still several major biological questions about this family that need to be addressed: What are the molecular mechanisms underlying the high oil content of Juglandaceae nuts? What is the genetic background of differences among individuals in the same species? Such questions could have profound commercial implications. For instance, due to the naked terminal buds of Chinese hickory and its specific climate needs, its cultivation range is geographically restricted8,9. Breeding stress-tolerant and vigorous Juglandaceae tree varieties is a long-term objective in the research community. Utilizing genomic data to understand the mechanisms of plant development and stress responses could help in breeding.
Recently, various genomic databases have been constructed due to the rapid development of plant genome sequencing10,11. However, to date, no specific database is available for walnut research. Many genomic data about the Juglandaceae family12,13,14 are accessible in public databases, such as GigaDB15 and NCBI16; however, most of these genomic data are unprocessed and decentralized. The data are neither functionally annotated and clustered in gene families nor preformatted for searching. Scientists have to process the data themselves after downloading them from public databases. However, processing these data is a challenge, especially for those without a bioinformatics background. Hence, these databases are among the least useful tools for the scientific community.
As mentioned above, we believe it is necessary to establish a shared resource center that integrates genomic data and germplasm (phenotype) resources of the Juglandaceae family. We built a comprehensive database for the Juglandaceae family, “Portal of Juglandaceae” (PJU, www.juglandaceae.net), that integrates their genomes, transcriptomes, and phenotypic data. We then mined, analyzed, and clustered these data appropriately to allow researchers to utilize the data more efficiently. Most importantly, the PJU has a user-friendly web interface for the scientific community. The PJU is also integrated with numerous practical bioinformatics tools that help researchers and users search, browse, or retrieve desired information from the portal. We hope, in time, that the PJU will become a comprehensive genomic data platform for the scientific community, especially for the study of Juglandaceae.
Construction of the PJU
Acquisition of genomic and transcriptomic data
Our colleagues previously reported the reference genomes of Chinese hickory (C. cathayensis) and pecan (C. illinoinensis)17. Reference genome, general feature format (GFF), coding sequence (CDS), protein sequence (PEP), and RNA expression data sets were obtained from the supporting data in the GigaScience database15.
The reference genomes of six Juglans and one Pterocarya species (J. cathayensi, J. hindsii, J. microcarpa, J. nigra, J. regia, J. sigillata, P. stenoptera) in the PJU were obtained from a previous study under Bioproject PRJNA44570418. The types of genomic data mentioned above were also downloaded from NCBI. A summary of genomic data currently available in the PJU is presented in Table 1.
The transcriptome expression data of the Juglandaceae family were recalculated from fragments per kilobase of exon model per million mapped fragments (FPKM) to transcripts per million (TPM) to present the relative transcript expression levels appropriately19.
Acquisition of phenotypic and physiological data
Germplasm resources are formed through thousands of years of natural evolution under different ecological conditions. They contain various potential genes and are important foundations for plant systematics research. Our group has been studying pecan germplasms from China and the United States for years. We have investigated essential features, such as flower phenology, fruit morphological indexes, fatty acids, and amino acid composition, in our pecan germplasm resources center. All the above data sets were converted to visual charts and are presented in the PJU.
Gene annotation, family, and synteny search
In attempts to decipher the unknown functions of tens of thousands of protein CDSs in individual genomes, researchers often need to perform comparisons using a variety of databases to make inferences on the sequences’ possible functions. InterProScan20 was used (with local mode and full analysis parameters) to analyze the protein domain functions of Juglandaceae family genes using searches against the InterPro and Pfam databases21. The predicted relevant domain and site results were classified according to HMM models using default parameters. Each gene was annotated with Gene Ontology (GO)22 terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG)23 to understand its function and location, respectively, in biochemical pathways.
The transcription factor families of Juglandaceae species were identified by searching against the Plant Transcription Factor Database24 using HMM models from the Pfam database (version 31.0)21. The HMMbuilder in the HMMER toolkit25 was also used (with default parameters) to assist in the identification process when an HMM model was not available.
Because of the close phylogenetic relationships among Juglandaceae species, there may be many homologous gene blocks in their genomes26. The Python version of MCScan27 (https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)) was used to find homologous gene blocks in genomes of the Juglandaceae family. The CDSs and coordinate files of nine species were extracted, and then the syntenic gene blocks between multiple genomes were obtained using the jcvi.compara module of Mcscan with the minspan parameter set at 30 genes.
BLAST and JBrowse modules, flanking sequence finder
BLAST28, one of the most popular bioinformatics tools for sequence similarity searches, was installed in the PJU. This module supports FASTA file dragging and multiple searches against preformatted genomes. JBrowse29, built using JavaScript and HTML5, was embedded in the PJU to browse and visualize the desired genomes. In this module, smooth scrolling and scaling visualization from the genome to nucleotide base level is supported.
With the two tools described above, researchers can easily find homologous genes Juglandaceae species. To find promoters, we utilized GFF and genome data and developed the Flanking Sequence Finder, which can help researchers find the flanking sequence of a specific gene and the exon and intron sequences.
Data integration and website construction
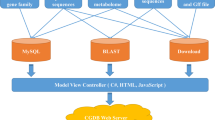
We created and deployed a user-friendly website for the PJU to make it easy for the scientific community to use the platform. The web pages are set up on the Huawei cloud server, one of the safest and most stable cloud service providers in the world. The server runs on the Ubuntu Linux system, uses Nginx as a web server and is also a protector of the PJU web application. The PJU data sets are stored in a MySQL database. To date, nine genomes, 488,299 PEPs, 1,792,549 GO annotations, and 750 miRNAs (Table 2) have been downloaded, analyzed, and organized in the MySQL database. We used Flask (a Python microframework) to fit the database web application into a single Python project. The PJU utilizes the Python program to handle data analysis and interactive tasks. The PJU web pages are built using HTML and Bootstrap and were designed to be user-friendly to researchers (Fig. 1). Moreover, useful bioinformatics tools (including JBrowse, BLAST, and others) are also bundled with genomic data sets in the database for researchers.

In short, processed genomic data are stored in a MySQL database on the server. A Flask framework-based website was built for visitors to utilize and download our data
Utilization of the PJU
The portal’s homepage
A clear and completely displayed homepage of the PJU (Fig. 2) has been built. Currently, the homepage includes five main parts: navigation bars, species galleries, tool sets, brief introductions, and other modules. The navigation bar (Fig. 2a), at the top of the homepage, consists of seven labels: Home, Introduction, Species, Tools, Download, Community, and Help. Under the navigation bar is the species gallery (Fig. 2b) of the PJU showcasing nine species. A tool set (Fig. 2c) containing BLAST, JBrowse, and several other practical search tools is on the right side of the species gallery. Below the species gallery and tool set, a brief introduction (Fig. 2d) of the PJU is given. At the same time, other useful information, including news, citations, and global visitors, is displayed at the bottom of the homepage (Fig. 2e).

The PJU homepage contains five components: a navigation bar, b species gallery, c tool set, d brief introduction, and e news, citation and visitors track. Visitors can browse for desired information using components
A brief introduction to the Juglandaceae family
In the four parts of the Introduction module, we educate visitors about the Juglandaceae family. The first part describes the origin of the Juglandaceae family. We investigated paleontological and phylogenetic studies on the Juglandaceae family and summarized them in detail in the web pages30,31,32. The second part is for taxonomy. We constructed a tree of the taxonomic status and position of Juglandaceae within the angiosperms and present important economic species of the Juglandaceae family. In the third part, we detail the economic value of the nine existing species in the Juglandaceae database. The fourth section lists the published literature on the Juglandaceae genome study and provides links to the full-text downloads.
In the Species module, we give basic introductions to the nine sequenced species and statistics on genome sequencing and provide users with links to BLAST, JBrowse, searches, and downloads.
The PJU search kits
We have developed a range of search tools through genomic data mining, including gene, transcription factor, gene family, synteny, miRNA, and pathway searches. A full map of search tools is presented in Fig. 3.

Scientists can use BLAST and JBrowse to find the homologous gene and its position and then use Gene Search to obtain related information. In addition, scientists can also utilize six search functions to obtain the desired information (left diagram). The DNA or protein sequences of the query gene set in FASTA format are also available for download
On the PJU, scientists can find homologous genes or a desired gene in the Juglandaceae family by utilizing the BLAST tool (Fig. 4a). Scientists can also find the specific location of the desired gene in the genome with the help of JBrowse (Fig. 4b). From the search results of BLAST and JBrowse, scientists can enter the gene’s identifier to search for information about the gene family, KEGG annotation, GO annotation (with links to the corresponding annotation database provided for more information), nucleotide coding and PEPs (FASTA format), and gene expression data based on TPM (Fig. 5).

Scientists can search for Juglandaceae homologous genes by using a the BLAST function. Scientists are also able to visualize the location of the query gene in the genome with the help of b JBrowse

The Portal of Juglandaceae (PJU) has a set of search tools that help scientists look for desired information. Scientists can enter the desired gene identifier, which is obtained from BLAST search results, to view a a report that contains the gene family, KEGG and GO annotations, nucleotide and protein sequences, and gene expression data based on TPM values. Scientists can search against b the gene family or c transcription factor database by entering a specific gene family or transcription factor name to search for related information. The PJU will return d a gene list that links to both e the FASTA file for viewing/downloading and detailed information, as mentioned above
Additionally, we also provide a feature to search for flanking sequences using the Flanking Sequence Finder module. With this tool, scientists can easily identify key regulatory elements and clone promoters.
The gene family search (Fig. 5b) and transcription factor search (Fig. 5c) functions are all based on specific gene search functions. The scientist selects the species and enters the transcription factor family or gene family name. After the search is conducted, the database returns a list of corresponding gene collections to the user (Fig. 5d). Scientists can click the gene ID, which is linked to detailed information (Fig. 5a) and the FASTA file (Fig. 5e).
The functionality of the syntenic gene block search is based on Mcscan27 software. We searched and organized the syntenic gene blocks for the whole genomes of multiple Juglandaceae species. Scientists can enter the gene ID and flanking gene numbers to view the collinear portion of the gene block against other species’ genomes (Fig. 6).

Scientists can look for the syntenic genes of Juglandaceae by a entering a gene identifier and deciding the number of flanking genes to be presented in the Synteny Browse Search. The PJU will return b a table of syntenic gene blocks between Juglandaceae species to scientists
In the pathway module, we selected important metabolic pathways from the KEGG database and identified some of the key genes in Arabidopsis thaliana. Scientists can find the sequences in TAIR33 http://arabidopsis.org via the link and BLAST the sequences in our database.
Finally, we also provide miRNA data for two species (C. cathayensis and C. illinoinensis), and the corresponding precursor sequences in the Juglandaceae family can be found by inputting the miRNA family name.
Juglandaceae breeding
For the breeding module, we used the existing germplasm repository of our campus to build a phenotype resource library. We focused on the observation and classification of the morphological changes in the pecan inflorescence, pollen vigor, and stigma receptivity between different cultivars. Additionally, we measured several important traits related to nut morphology and fatty acid components.
Although pecan (C. illinoinensis) is a monecious tree with diecious flowers, most of its varieties are actually expressed as diecious, which means that they can avoid self-pollination, thus improving the quality, yield, and genetic diversity of the nut34. Our research team conducted continual germplasm surveys focusing on flowering and nut characteristics to understand the flower phenological and flower bud differentiation mechanisms.
The phenotypes of wild and domesticated germplasm resources of C. cathayensis are still being sampled and collated. We will share these data with the community once they are ready.
Download and community modules
The Download module provides the genome assembly, CDS and PEPs, annotation data, and miRNA downloads in FASTA and GFF3 formats. Researchers can also directly access data from the chloroplast genome. Data on the expression levels of different genes during three developmental stages17 (the early and fully extended stages of cotyledon development and the fully mature stage of embryos) of plant growth and development are presented in CSV format.
For the Community module, we collected the literature on Juglandaceae research. Scientists can search this literature by date, species, or research type.
Conclusion and future developments
With the rapid development of sequencing technology and corresponding bioinformatics software, an increasing number of plants in the Juglandaceae family have entered the era of research with high-throughput data. How to effectively use and mine this valuable data on a benchtop sequencer will become the focus of modern biological research. We conducted genomic data mining and established the PJU. The PJU aims to be not only a genomic information database but also a user-friendly portal that integrates several useful genomic datasets under a simple website framework.
When new higher quality genomic data emerge, we will collect and analyze them and immediately release them into the PJU. We hope that the Portal of Juglandaceae will be a hub for future genomic research on the Juglandaceae family.
References
Manos, P. S. & Stone, D. E. Evolution, phylogeny, and systematics of the Juglandaceae. Ann. Mo. Bot. Gard. 88, 231 (2001).
Grauke, L., Wood, B. & Harris, M. Crop vulnerability: Carya. HortScience 51, 653–663 (2016).
Chen, F. et al. Genomics: cracking the mysteries of walnuts. J. Genet. 98, 33 (2019).
Burtin, P., Jay-Allemand, C., Charpentier, J. P. & Janin, G. Natural wood colouring process in Juglans sp. (J. nigra, J. regia and hybrid J. nigra 23 × J. regia) depends on native phenolic compounds accumulated in the transition zone between sapwood and heartwood. Trees—Struct. Funct. 12, 258–264 (1998).
Zhu, C., Deng, X. & Shi, F. Evaluation of the antioxidant activity of Chinese Hickory (Carya cathayensis) kernel ethanol extraction. Afr. J. Biotechnol. 7, 2169–2173 (2008).
Bolling, B. W., Chen, C. Y. O., McKay, D. L. & Blumberg, J. B. Tree nut phytochemicals: composition, antioxidant capacity, bioactivity, impact factors. A systematic review of almonds, Brazils, cashews, hazelnuts, macadamias, pecans, pine nuts, pistachios and walnuts. Nutr. Res. Rev. 24, 244–275 (2011).
Miraliakbari, H. & Shahidi, F. Antioxidant activity of minor components of tree nut oils. Food Chem. 111, 421–427 (2008).
Grauke, L. J., Wood, B. W. & Harris, M. K. Crop vulnerability: Carya. HortScience 51, 653–663 (2016).
Yang, J. et al. Comparison of phenolic compounds, tocopherols, phytosterols and antioxidant potential in Zhejiang pecan [Carya cathayensis] at different stir-frying steps. LWT - Food Sci. Technol. 62, 541–548 (2015).
Chen, F. et al. Genome sequences of horticultural plants: past, present, and future. Hortic. Res. 6, 112 (2019).
Chen, F. et al. The sequenced Angiosperm Genomes and Genome Databases. Front. Plant Sci. 9, 1–14 (2018).
Wu, G. A. & Gmitter, F. G. Novel assembly strategy cracks open the mysteries of walnut genome evolution. Hortic. Res. 6–7 (2019). https://doi.org/10.1038/s41438-019-0143-5
Mccoy, R. M. & Utturkar, S. M. The origin and biosynthesis of the naphthalenoid moiety of juglone in black walnut. Hortic. Res. (2018). https://doi.org/10.1038/s41438-018-0067-5
Zhu, T. et al. Sequencing a Juglans regia × J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 6, 1–16 (2019).
Sneddon, T. P., Li, P. & Edmunds, S. C. GigaDB: announcing the GigaScience database. Gigascience 1, 11 (2012).
Agarwala, R. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19 (2016).
Huang, Y. et al. The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. Gigascience 8, giz036 (2019).
Stevens, K. A. et al. Genomic variation among and within six Juglans species. G3 Genes|Genomes|Genet. 8, 2153–2165 (2018).
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A. & Dewey, C. N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500 (2009).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Carbon, S. et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019).
Kanehisa, M. & Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 1–8 (2019). https://doi.org/10.1002/pro.3711
Pérez-Rodríguez, P. et al. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, 822–827 (2009).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011).
Van De Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 1–14 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 1–9 (2009).
Buels, R. et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 1–12 (2016).
Blokhina, N. I. Fossil wood of the Juglandaceae: some questions of taxonomy, evolution, and phylogeny in the family based on wood anatomy. Paleontol. J. 41, 1040–1053 (2007).
Manos, P. S. et al. Phylogeny of extant and fossil juglandaceae inferred from the integration of molecular and morphological data sets. Syst. Biol. 56, 412–430 (2007).
Manchester, S. R. Early history of theJuglandaceae. Plant Syst. Evol. 162, 231–250 (1989).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, 1202–1210 (2012).
Thompson, T. E. & Romberg, L. D. Inheritance of heterodichogamy in pecan. J. Hered. 76, 456–458 (1985).
Acknowledgements
The study was supported by grants from the National Natural Science Foundation of China (31570666), Major Science and Technology Projects of Zhejiang Province (2018C02004), Major Scientific and Technological Projects for the Selection of New Varieties of Agriculture in Zhejiang Province (2016C02052-13), and National Key R&D Program of China (2018YFD1000604).
Author information
Authors and Affiliations
Contributions
Z.J.W. and W.L.G. conceived and designed the research; J.L. conducted phenotypic experiments; J.Q.H. provided genomic and transcriptomic data sets of Carya cathayensis and Carya illinoinensis; W.L.G. and J.H.C. designed the tools and obtained and analyzed the data; W.L.G. organized and constructed the portal architecture; W.L.G. and K.-J.L. designed the layout of the web pages; W.L.G. wrote and K.-J.L. revised thoroughly the manuscript. All authors read, commented on, and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, W., Chen, J., Li, J. et al. Portal of Juglandaceae: A comprehensive platform for Juglandaceae study. Hortic Res 7, 35 (2020). https://doi.org/10.1038/s41438-020-0256-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-0256-x
This article is cited by
-
Morphological, pomological, and biochemical evaluation of several superior walnut (Juglans regia L.) genotypes
Genetic Resources and Crop Evolution (2024)
-
Whole genome based insights into the phylogeny and evolution of the Juglandaceae
BMC Ecology and Evolution (2021)
