
Abstract
Cytoplasmic male sterility (CMS) is an important tool for producing F1 hybrids, which can exhibit heterosis. The companion system, restorer-of-fertility (Rf), is poorly understood at the molecular level and would be valuable in producing restorer lines for hybrid seed production. The identity of the Rf gene in Capsicum (pepper) is currently unclear. In this study, using bulked segregant RNA sequencing (BSR-seq), a strong candidate Rf gene, Capana06g002866, which is annotated as a NEDD8 conjugating enzyme E2, was identified. Capana06g002866 has an ORF of 555 bp in length encoding 184 amino acids; it can be cloned from F1 plants from the hybridization of the CMS line 8A and restorer line R1 but is not found in CMS line 8A. With qRT-PCR validation, Capana06g002866 was found to be upregulated in restorer accessions compared to sterile accessions. The relative expression in flower buds increased with the developmental stage in F1 plants, while the expression was very low in all flower bud stages of the CMS lines. These results provide new insights into the Rf gene in pepper and will be useful for other crops utilizing the CMS system.
Similar content being viewed by others


Introduction
Capsicum species serve as popular vegetables and spices around the world1. Capsicum annuum is the most widely grown among the five domesticated species (C. annuum, C. baccatum, C. chinense, C. frutescens, and C. pubescens)1. As with other crops, the use of F1 hybrid seed can greatly improve yield, resistance, and quality in peppers. The production of pepper hybrid seed requires manual emasculation, which increases the cost of seed production, and the purity of hybrid seeds produced is sometimes low.
The use of cytoplasmic male sterility and the corresponding fertility restorer is the most valuable system for exploiting hybrid vigor; it saves labor and time because it does not require manual emasculation and ensures the purity of hybrid seeds2,3. It has been proven that CMS is maternally inherited and is determined by mitochondrial genes resulting from chimeric ORF rearrangements that disturb the normal development of pollen4. However, Rf genes in the nucleus can override the expression of sterility or prevent the accumulation of CMS-specific gene products and in turn reverse the CMS phenotype5,6,7,8,9,10. It has also been suggested that Rf genes may be involved in the detoxification of acetaldehyde produced by ethanolic fermentation during pollen development11. At present, Rf genes have been cloned in a few species, such as rice (Oryza sativa)12,13, radish (Raphanus sativus)14,15,16,17,18, sorghum (Sorghum bicolor)19, and petunia (Petunia × atkinsiana)6. Among these Rf genes, most encode pentatricopeptide repeat (PPR) proteins8,20,21. Other types of Rf genes have also been cloned in rice and maize (Zea mays). For example, the Rf2 genes in maize and rice encode an aldehyde dehydrogenase protein and a glycine-rich protein, respectively11,22, and the Rf17 gene in rice encodes a protein of unknown function22. Thus, different types of Rf genes exist in plants.
In pepper, two CMS genes, atp6 and orf456, have been studied23,24. It has also been reported that orf507 from the alternate stop codon of orf456 can inhibit the formation of microspores and result in CMS in pepper25. Furthermore, two CMS-specific sequence-characterized amplified region (SCAR) markers, coxII and atp6, have been developed from the sequences flanking orf456 and atp6-2, respectively26. In addition, another molecular marker of S-cytoplasm, SCAR130, was reported to be more reliable than the previous markers27.
However, no Rf gene has been reported in pepper thus far. Some studies have reported that fertility restoration is regulated by one major gene. However, several cases also indicate that more complex mechanisms for fertility restoration may exist28,29,30. Additionally, partial fertility restoration of male sterility with multiple haplotypes also exists31,32,33.
As a highly conserved 76-amino-acid polypeptide, ubiquitin can be conjugated to protein substrates through ubiquitination34. At the initial step of ubiquitination, ubiquitin is activated by a Ub-activating enzyme (E1) through the formation of a covalent thioester bond between a cysteine residue in the E1 active site and the C-terminal end of the ubiquitin, which requires the participation of ATP. The activated ubiquitin is then transferred from E1 to a Ub-conjugating enzyme (E2) to form the E2-ubiquitin complex. Then, both the E2-bound ubiquitin and the protein substrate are connected to each terminal of a Ub ligase (E3) to form a substrate-E3-E2-ubiquitin complex. Subsequently, with the catalytic action of E3, ubiquitin is transferred to the protein substrate and forms a monoubiquitinated substrate, accompanied by the release of E2 and E3. A monoubiquitinated substrate can also have additional ubiquitin molecules added to it one by one via Lys48 of the ubiquitin molecule to form a polyubiquitinated substrate34,35. The ubiquitin-proteasome system (UPS) is the main degradation pathway of proteins in eukaryotic cells and plays a vital role in many biological processes36,37.
In addition to ubiquitin, there are some ubiquitin-like proteins (UBLs), including small ubiquitin-like modifiers (SUMOs), that act similarly to ubiquitin38. Among these UBLs and SUMOs, NEDD8 is the most similar to ubiquitin at both the sequence and secondary structure levels, with approximately 60% amino acid similarity identity38,39,40,41. Analogous to ubiquitination, the process of NEDD8 conjugation to a substrate is called neddylation and is catalyzed in a three-enzyme cascade (E1, E2, and E3)42,43. Neddylation plays fundamental roles in signal transduction, cell division, morphogenesis, and embryogenesis43,44,45,46.
By sequencing transcripts directly, the RNA-Seq method has considerable advantages for evaluating allele-specific expression, detecting novel transcripts and providing other genome-wide information from high-throughput sequencing technologies47. Separately, bulked segregate analysis (BSA) was developed as an efficient way to rapidly develop molecular markers for specific traits or target gene loci48. BSR-seq, a new method combining BSA and RNA-seq, is used not only to map traits on chromosomes but also to provide gene expression information, including information for the genes within the mapping regions49,50,51. BSR-Seq identifies differentially expressed genes (DEGs) between the two extreme-phenotype pools within the mapping interval, and the function of the candidate DEGs can be annotated by bioinformatics analysis52. Using the BSR-seq method, many candidate DEGs have been identified in several plant species, such as wheat (Triticum aestivum)53, cabbage (Brassica oleracea)54, and onion (Allium cepa)55.
In this study, two extreme-phenotype gene pools, a sterile pool, and a restorer pool, were constructed from F2 individuals of a cross between the sterile line and fertility restorer line. First, through the BSR-seq method, an approximately 16.8 M mapping region was selected, and five genes were detected to be commonly upregulated in the restorer pool and restorer line. Subsequently, only one gene, Capana06g002866, annotated as a NEDD8 conjugating enzyme E2, was cloned and chosen as a strong candidate Rf gene. The results provide new insights into the molecular cloning of a candidate Rf gene for CMS in pepper.
Results
SNP calling and filtering
Through SNP calling, more than 165,500 SNPs were detected in 8A, R1, SP, and RP (Table 1). Notably, the number of SNPs was higher in the restorer accessions (R1 and RP) than in sterile accessions (8A and SP) regardless of subcategory. In addition, the number of homozygous SNPs far outweighs that of heterozygous SNPs in all samples.
To ensure the accuracy of subsequent analysis, the SNPs were filtered. First, degenerate SNPs were filtered, and then the sites supported with fewer than three reads were removed. Finally, the sites where SNPs differed between the extreme-phenotype pools and its corresponding parent were filtered out. After that, the high quality and credible SNPs were retained. On the basis of these reliable SNPs, a total of 62,629 polymorphic sites between the two extreme-phenotype pools were identified, which were then used to perform the association analysis (Table 2). The SNPs were distributed on all 12 chromosomes, and some were not assigned to any chromosome (chromosome00) (Fig. 1). The chromosomes with the most SNPs were chromosome03 and chromosome00, 11.71% and 11.64%, respectively. The chromosomes with the fewest SNPs were chromosome11 and chromosome04, with 5.11% and 5.24%, respectively.
Preliminary mapping of the Rf gene
Through the ED algorithm, only one single peak was observed to exceed the threshold significantly, on the end of chromosome06 (Fig. 2). The median + 3SD of all site fitted values, which was 0.033421494897978, was taken as the correlation threshold for analysis. An interval above the threshold was screened as the mapping interval where the Rf gene was located. This indicated that the Rf gene was mapped to an interval of 16.8 Mbp (Chr06: 199389022-216191732), close to the end of chromosome06 (Fig. 2). In addition, there was more than 76% identity between the identified fertility region and KASP genotyping of KS18 and KS22 for the 72 F2 individuals, which indicated that the region mapping was reliable. The mapping region was further narrowed to approximately 5.1 Mb (Chr06: 210576870–215685280) according to two SNP locations derived from the KASP markers KS18 and KS22.

Each part of the pie chart represents a chromosome and the percentage of SNPs

a The association peaks shown on all the chromosomes. b Magnification of the peak showing only chromosome 6
In this mapping interval, 199 genes, from Capana06g002727 to Capana06g002990 were detected, of which 93, 104, 105, and 112 genes were detected in 8A, R1, SP, and RP, respectively, and 84 genes were common to all pools (Fig. 3). Between the two extreme-phenotype pools, there were eight genes upregulated and two genes downregulated in the RP compared to the SP. To increase the range limit of candidate genes, it was necessary to identify the common upregulated and downregulated genes. Between the two parent lines, nine genes were upregulated and seven genes were downregulated in R1 compared to 8A. Interestingly, five genes (Capana06g002839, Capana06g002848, Capana06g002866, Capana06g002871, and Capana06g002913) were upregulated in both R1 and RP compared to 8A and SP, and one gene (Capana06g002754) was downregulated in both (Fig. 4, Table 3). It was also interesting that a gene (Ca06g002814) encoding a PPR protein was detected in the mapping region. The FPKM and log2FC values of these genes are listed in Table 3.

a The number of genes detected in all and different materials. b Venn analysis of the genes detected in the four materials

a Venn analysis of upregulated genes in common. b Venn analysis of the downregulated genes in common. Parents indicate the differentially expressed genes between two parent lines, pools indicate differentially expressed genes between two extreme pools, overlap indicates the differentially expressed genes in common, and number indicates the number of differentially expressed genes
Screening and expression of the strong candidate Rf gene
The results of the candidate gene analysis comparing 8A and F1 showed that Ca06g002866 amplified the target fragments in F1 but not in 8A, and Ca06g002839 showed a very weak band in F1 but not in 8A. While four genes (Capana06g002848, Capana06g002871, Capana06g002754, and Capana06g002814) produced fragments in both 8A and F1, one gene, Ca06g002839, did not produce any bands in either F1 or 8A (Fig. 5). Therefore, we chose Ca06g002866 as the best candidate Rf gene in pepper. Sequencing results showed that the ORF length of Capana06g002866 is 555 bp, the same as the reference gene sequence, and encodes 184 amino acids. Functional annotation indicates that Capana06g002866 encodes a Ubc12-like NEDD8-conjugating enzyme.

The expression of candidate genes differ between the flower buds of 8A and F1. The left collumn means the name of candidate genes, the middle electrophoresis shows the results of RT-PCR of candidate gene in the flower buds of 8A and F1, and the right collumn indicates the relative expressin of candidate genes in the flower buds of F1 compared to that of 8A by qRT-PCR method
Subsequent qRT-PCR confirmed the differential expression of the candidate genes, and the results of qRT-PCR were identical to the sequencing results, which indicated that the expression of Capana06g002866 was obviously upregulated in restorer accessions compared to sterile accessions (Fig. 6a). Furthermore, the expression pattern indicated that the relative expression of Capana06g002866 was very low across different flower developmental stages in 8A, while it varied dramatically in the F1 plants (Fig. 6b). The relative expression began to increase at stage III and quickly peaked at stage IV (Fig. 6b).

a qRT-PCR validation in the sequenced accessions. b Analysis of relative expression in different developmental stages in 8A and F1
Discussion
The utilization of the CMS/Rf system is one of the most effective ways to produce F1 hybrids. The Rf genes encode different proteins that restore the fertility of CMS through different mechanisms, and the identity of the gene (or genes) involved in fertility restoration in pepper, as well as the mechanism through which it works, is still unclear. Clarification of the mechanism of fertility restoration for CMS would be helpful for accelerated breeding of restorer lines.
The BSR-seq method provides a powerful approach for selecting candidate genes53,54,55. In this study, BSR-seq was used to map a region of 18.6 Mbp on chromosome 06 that restores fertility. Using common upregulated DEG analysis, the five most upregulated DEGs were selected for further analysis. We examined the sequence counts and FPKM in the restorer and sterile pools. These values should be zero or very low in sterile accessions and higher in fertile accessions (Table 3). We further amplified the candidate genes within the mapping region and found that only Capana06g002866 could be cloned in F1 plants but not in CMS line 8A (Fig. 5), which was consistent with the requirements of the Rf gene. In addition, Capana06g002866 was upregulated in restorer accessions and showed very little expression in sterile accessions, which was further validated by the qRT-PCR results. It should be noted that Capana06g002866 is predicted to be a NEDD8 conjugating enzyme. Therefore, Capana06g002866 is the most likely candidate for involvement in the restoration of fertility in pepper.
The NEDD8 conjugating enzyme is the second key enzyme in the three-enzyme cascade process of neddylation42,43. Neddylation plays fundamental roles in signal transduction, cell division, morphogenesis, and embryogenesis43,44,45,46. Neddylation is also involved in pollen development, and it has been demonstrated that the NEDD8 ligase DCN1 (DEFECTIVE IN CULLIN NEDDYLATION 1) is very important in pollen development and pollen tube growth of tobacco46. Additionally, it has been reported that several genes related to ubiquitin ligase were upregulated in fertile anthers compared to CMS lines in pepper56. This indicates that neddylation is involved in pollen production and could be involved in the regulation of male fertility in pepper. Another example of ubiquitin involvement in pollen development is that the ubiquitination level of the 80 kDa protein produced in male sterile flower—in wheat decreased significantly with the development stage and reached the lowest level during the trinuclear stage57.
Ubiquitination function depends on the enzymatic cascade of E1, E2, and E3. The E3 ligase is a very important enzyme for conjugating the ubiquitin molecule to its substrate. HECT, RING finger, U-box, and PHD finger are the four classes of E3 ubiquitin ligases identified to date58. Cullin RING E3 ubiquitin ligases (CRLs) are the most prominent E3 type among the four classes59. Cullins are the main component of CRLs, and the activation of CRLs depends on the neddylation of cullin37,60,61,62. Notably, one of the genes upregulated in restorer lines in this study (Capana06g003066) encodes a cullin protein. With catalysis by E3, the substrates are first modified by a single ubiquitin molecule, and then other ubiquitin molecules are added one by one to form polyubiquitination34,35. The polyubiquitinated substrate is then recognized by the 26 S proteasome and degraded with the release of single ubiquitin molecules34,63,64,65. The ubiquitin-mediated proteasome system (UPS) relies on this well-known posttranslational modification (PTM) of proteins and is the major degradation pathway of cellular proteins in general. The UPS plays an important role in cell cycle progression, apoptosis, stress response, and growth and development processes in eukaryotes35,66,67. It has been reported that ubiquitination and UPS are involved in acrosome biosynthesis and sperm tail formation in human and animal spermatogenesis, UPS plays an important role in the degradation of organelles and excess protein in sperm metamorphosis, and abnormal regulation of UPS leads to sperm deformity68,69,70,71.
It can be inferred that the NEDD8-conjugating enzyme gene may restore the fertility of CMS through the activation of CRLs, which in turn promote ubiquitination in the restoration process. Ubiquitination is known to tag substrates for degradation by the 26 S proteasome. Neddylation facilitates ubiquitination by activating CRLs through modification on cullins, which are known to be important components of E3 for ubiquitination72. Because of this relationship, the increase in NEDD8-conjugating enzyme expression in R1 and RP could be interpreted as an increase that facilitates degradation. This would make sense in the context of fertility restoration if the substrate is the pollen-aborting mitochondrial ORF being tagged for degradation.
In addition, recent research clarified that neddylation is also involved in the DNA damage response and DNA repair process37,73,74. DNA damage is detected in the nuclei of pollen mother cells (PMCs) of CMS plants in pepper56. The damage may cause abnormal programmed cell death (PCD)75. Disturbed PCD in the tapetum will lead to a CMS phenotype76. These observations seem to indicate that neddylation may restore fertility by repairing DNA damage. Whether neddylation could be involved in fertility restoration through UPS or DNA repair requires further study.
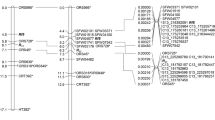
It is interesting that the candidate Rf genes in our study and the previous studies were all located on the terminus of chromosome 6, such as CaPPR_4677,78, CaRf03279, and Capa06g00286680 (Fig. 7). However, the candidate Rf gene in our study encoded a NEDD8 conjugating enzyme, while the other candidates were mainly PPR proteins. It is worth mentioning that the majority of Rf genes encode PPR proteins8,20,21. The PPR genes are members of a protein family that is exceptionally large in plants, and pepper is no exception, with more than 550 putative PPR genes identified77. Some attempts have been made to find pepper Rf genes with a PPR structure77,78. In this study, it is interesting that a gene, Capana06g002814, is a predicted PPR protein that was mapped to the same region related to the restorer-of-fertility for CMS in pepper. However, Capana06g002814 was downregulated in fertility restorer accessions compared to cytoplasmic male sterile accessions and could not be detected in R1 because the sequence number and FPKM were zero. Logically, the Rf gene should be upregulated in restorer lines and not expressed in CMS lines. When compared to previously identified PPR genes presumed to be related to restorer-of-fertility function (CaPPR1 and CaPPR6)73, Capana06g002814 showed 86% and 84% similarity, respectively. Because of the nature of PPRs as a protein family are similar in structure, it is difficult to say whether this 84–86% similarity in structure indicates any similarity in function. However, this PPR protein consisted of only nine motifs, while Rf-like PPR proteins are generally approximately 15–20 motifs long20, with the CaPPR1 and CaPPR6 genes consisting of 14 PPR motifs78. Thus, it is more likely coincidental that this PPR gene appears in the mapping region than that it plays an actual role in fertility restoration.

The location of several Rf candidate genes on chromosome 6 of the reference genome of Zunla1
Materials and methods
Sample, transcriptome sequencing, and DEGs
The CMS line 8A (C. annuum L.), the restorer line R1 (C. annuum L.), and two extreme pools, a sterile pool (SP) and a restorer pool (RP), were described in our previously published articles81. The methods of transcriptome sequencing, quantification of gene expression levels and DEG analysis were the same as in the previous articles81.
SNP calling and filtering
For transcript sequencing, the reads of each sample were mapped to the reference genome using the comparison software STAR (https://github.com/alexdobin/STAR/releases), and the SNP sites were found through the SNP calling flow of GATK (https://www.broadinstitute.org/gatk/index.php) for RNA-seq. The quality value of recognition and the depth of sequencing affect the reliability of SNPs, so to obtain the most reliable SNPs, elementary screening of SNPs was carried out according to the following criteria. First, no more than 3 continuous mismatched SNPs within a 35 bp range were permitted. Second, the quality value of the SNP after the standardization for sequence depth was greater than 2.0. In addition, to identify the SNP loci differing between RP and SP for association analysis, discrepant-type SNP loci between RP and R1, as well as SP and 8A, were filtered out, and then SNP consistent-type loci between RP and SP were filtered out.
Association analysis
The Euclidean distance (ED) algorithm calculated the region linked to the target gene82. In this algorithm, the depth differences of SNPs between the extreme fertile pool and extreme sterile pool calculated the ED values between two extreme pools according to the following formula:
In the formula, the higher the ED value is, the greater the difference in the SNP between the two extreme pools. The letters A, C, G, and T represent the four corresponding bases. Theoretically, the ARP in the formula is the depth of base A in the extreme fertile pool, and ASP is the depth of base A in the extreme sterile pool. However, in practice, the difference in sequencing quantity between mixed pools will lead to the bias of ED results. To eliminate this error, the frequency of the base at each point instead of absolute depth was used to calculate the ED value in this project. In addition, the original ED value was raised to a power of 5 (ED5) to decrease the background noise.
According to the linkage principle, the SNP loci nearest the real association region will tend to differ between the two extreme pools. To control the false positives caused by the association of a single locus, fitting analysis is performed depending on the location information of the SNP in the reference genomes. The fitting correlation value for each SNP was the median of the 50 up- and downstream SNPs.
Validation of the mapping region
A KASP assay was used to further confirm the mapping region by examining the SNP flanking the restorer locus in the 72 F2 individuals of the cross of 8A and R1 (Table 4). Allele-specific primers for the KASP assay were designed with the online version of BatchPrimer3 V1.0 (https://probes.pw.usda.gov/batchprimer3/) and synthetized by Sangon Biotech (Shanghai) Co., Ltd. The primer pairs were composed of two forward primers, A1 and A2, and reverse primer C. The two allele-specific forward primers had the same primer sequence except for the targeted SNP at the 3′ end and their FAM and HEX-labeled tails (FAM tail: 5′-GAAGGTGACCAAGTTCATGCT-3′; HEX tail: 5′-GAAGGTCGGAGTCAACGGATT-3′), respectively. KASP assays were performed in 96-well plates with a 10 µl reaction mixture composed of 5 µl template DNA (30 ng µl−1), 2 µl of 2× KASP Master Mix, and 0.14 µl primer mixture according to the KASP genotyping chemistry user guide and manual (LGC Genomics, Shanghai, China). The KASP was performed on QuantStudio®5 (Applied Biosystems, US) as follows: an initial denaturation step at 94 °C for 15 min, followed by 10 touchdown cycles at 94 °C for 20 s and 61–55 °C for 60 s (dropping 0.6 °C per cycle), followed by 26 cycles at 94 °C for 20 s and at 55 °C for 60 s, and three additional cycles at 94 °C for 20 s and 57 °C for 60 s.
Cloning of candidate genes
First-strand cDNA was obtained using a synthesis kit for first-strand cDNA (Revert Aid Premium Reverse Transcriptase) (Thermo Scientific, EP0733). Pairs of primers (Table 5) for amplification of candidate genes were designed according to the reference sequences. The PCR conditions were as follows: denaturation at 95 °C for 5 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 45–60 °C for 30 s and extension at 72 °C for 50 s–2 min; and a final extension at 72 °C for 10 min. The target PCR product was purified and recovered using the AxyPrep Gel DNA Extraction Kit (Axygen, China) and cloned into the vector pMD19-T (TaKaRa, Dalian, China) for sequencing.
qRT-PCR analysis of Capana06g002866
The qRT-PCR primers (Capana06g002866-Fw-q: AGGGAAGATTGGAAACCTGTT; Capana06g002866-Rv-q: ATCTTTCAACTCATTGGCAGC) tested its relative expression, and CaActin (GenBank Accession: GQ339766.1) was chosen as an internal control (CaActin-Fw-q: TGCCTGATGGACAAGTTATTACC; CaActin-Rv-q: TGAGCACAATGTTACCGTAGAGG). qRT-PCR was carried out in a total reaction volume of 20 µL, including SG Fast qPCR Master Mix (High Rox) (2×) (BBI, B639273). qRT-PCR was performed with the following parameters: denaturation at 95 °C for 3 min, followed by 45 cycles of denaturation at 95 °C for 7 s, annealing at 57 °C for 10 s, and extension at 72 °C for 15 s on an ABI StepOne Plus Real-Time PCR System (Applied Biosystems, USA). At least three replicates were performed for all reactions.
Data availability
The raw sequences can be found in the Sequence Read Archive (trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi), and the temporary SRA accession number is PRJNA547768.
References
Bosland, P. W. & Votava, E. J. (eds). In Vegetable and Spice Capsicums, 2nd edn. (CAB International, Wallingford, 2012).
Kaul, M. L. H. (eds). In Male Sterility in Higher Plants (Springer-Verlag, Berlin, Heidelberg, 1988).
Havey, M. J. In Daniell H., Chase C., editors. Molecular Biology and Biotechnology of Plant Organelles (eds Daniell, H. & Chase, C.) 623–634 (Springer, Dordrecht, 2004).
Tuteja, R. et al. Cytoplasmic male sterility-associated chimeric open reading frames identified by mitochondrial genome sequencing of four cajanus genotypes. DNA Res. 20, 485–495 (2013).
Janska, H., Sarria, R., Woloszynska, M., Arrieta-Montiel, M. & Mackenzie, S. A. Stoichiometric shifts in the common bean mitochondrial genome leading to male sterility and spontaneous reversion to fertility. Plant Cell 10, 1163–1180 (1998).
Bentolila, S., Alfonso, A. A. & Hanson, M. R. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male sterile plants. Proc. Natl. Acad. Sci. USA 99, 10887–10892 (2002).
Wang, Z. et al. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18, 676–687 (2006).
Chase, C. D. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet 23, 81–90 (2007).
Kazama, T., Nakamura, T., Watanbe, M., Sugita, M. & Toriyama, K. Suppression mechanism of mitochondrial ORF79 accumulation by Rf1 protein in BT-type cytoplasmic male sterile rice. Plant J. 55, 619–628 (2008).
Colombo, N. & Galmarini, C. R. The use of genetic, manual and chemical methods to control pollination in vegetable hybrid seed production: a review. Plant Breeding 136, 287–299 (2017).
Cui, X., Wise, R. P. & Schnable, P. S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 272, 1334–1336 (1996).
Akagi, H. et al. Positional cloning of the rice rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeted PPR protein. Theor. Appl. Genet. 108, 1449–1457 (2004).
Komori, T. et al. Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J. 37, 315–325 (2004).
Brown, G. G. et al. The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J. 35, 262–272 (2003).
Deslorie, S. et al. Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep. 4, 588–594 (2003).
Koizuka, N. et al. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 34, 407–415 (2003).
Uyttewaal, M. et al. Characterization of Raphanus sativus pentatricopeptide repeat proteins encoded by a fertility restorer locus for Ogura cytoplasmic male sterility. Plant Cell 20, 3331–3345 (2008).
Wang, Z. et al. An insertion-deletion at a pentatricopeptide repeat locus linked to fertility transition to cytoplasmic male sterility in radish (Raphanus sativus L.). Mol. Breeding 25, 1–5 (2015).
Klein, R. R. et al. Fertility restorer locus Rf1 of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the collinear region of rice chromosome 12. Theor. Appl. Genet. 111, 994–1012 (2005).
Fujii, S., Bond, C. S. & Small, I. D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl Acad. Sci. USA 108, 1723–1728 (2011).
Dahan, J. & Mireau, H. The Rf and Rf-like PPR in higher plants, a fast evolving subclass of PPR genes. RNA Biol. 10, 1286–1293 (2013).
Hu, J. et al. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 19, 282–288 (2014).
Kim, D. S. & Kim, B. D. The organization of mitochondrial atp6 gene region in male fertile and CMS lines of pepper (Capsicum annuum L.). Curr. Genet. 49, 59–67 (2006).
Kim, D. H., Kang, J. G. & Kim, B. D. Isolation and characterization of cytoplasmic male sterility-associated orf456 gene of chili pepper (Capsicum annuum L.). Plant Mol. Biol. 63, 519–532 (2007).
Gulyas, G., Shin, Y., Kim, H., Lee, J. S. & Hirata, Y. Altered transcript reveals an orf507 sterility-related gene in chili pepper (Capsicum annuum L.). Plant Mol. Biol. Rep. 28, 605–612 (2010).
Kim, D. H. & Kim, B. D. Development of SCAR markers for early identification of cytoplasmic male sterility genotype in chili pepper (Capsicum annuum L.). Mol. Cells 20, 416–422 (2005).
Ji, J. J., Huang, W., Yin, Y. X., Li, Z. & Gong, Z. H. Development of a SCAR marker for early identification of S-cytoplasm based on mitochondrial SRAP analysis in pepper (Capsicum annuum L.). Mol. Breeding 33, 679–690 (2014).
Gulyas, G., Pakozdi, K., Lee, J. S. & Hirata, Y. Analysis of fertility restoration by using cytoplasmic male-sterile red pepper (Capsicum annuum L.) lines. Breeding Sci. 56, 331–334 (2006).
Novak, F., Betlach, J. & Dubovsky, J. Cytoplasmic male sterility in sweet pepper (Capsicum annuum L.) I. Phenotype and inheritance of male sterile character. Zeitschrift Pflanzenzuchtung 65, 129–140 (1971).
Wang, L. et al. QTL analysis of fertility restoration in cytoplasmic male sterile pepper. Theor. Appl. Genet. 109, 1058–1063 (2004).
Lee, D. H. Studies on Unstable Fertility of CGMS (Cytoplasmic Genetic Male Sterility) in Capsicum annuum L. Ph.D dissertation (Seoul National Univ. Seoul, Korea, 2001).
Lee, J., Yoon, J. B. & Park, H. G. Linkage analysis between the partial restoration (pr) and the restorer-of-fertility (Rf) loci in pepper cytoplasmic male sterility. Theor. Appl. Genet. 117, 383–389 (2008).
Min, W. K. et al. Identification of a third haplotype of the sequence linked to the restorer-of-fertility (Rf) gene and its implications for male sterility phenotypes in peppers (Capsicum annuum L.). Mol. Cells 25, 20–29 (2008).
Kleiger, G. & Mayor, T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 24, 352–359 (2014).
Glickman, M. H. & Ciechanove, R. A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428 (2002).
Yoshida, K. et al. Ubiquitin-activating enzyme E1 inhibitor PYR-41 retards sperm enlargement after fusion to the egg. Reprod. Toxicol. 76, 71–77 (2018).
Guan, J. & Zheng, X. NEDDylation regulates RAD18 ubiquitination and localization in response to oxidative DNA damage. Biochem. Biophys. Res. Commun. 508, 1240e1244 (2019).
Wang, Z., Zhu, W. G. & Xu, X. Ubiquitin-like modifications in the DNA damage response. Mutat. Res. Fund. Mol. Mech. Mutagen 803, 56–75 (2017).
Kamitani, T., Kito, K., Nguyen, H. P. & Yeh, E. T. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562 (1997).
Kumar, S., Tomooka, Y. & Noda, M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem. Biophys. Res. Commun. 185, 1155–1161 (1992).
Swatek, K. N. & Komander, D. Ubiquitin modifications. Cell Res. 26, 399–422 (2016).
Lammer, D. et al. Modification of yeast Cdc53p by the ubiquitin-related protein rub1p affects function of the SCFCdc4 complex. Gene Dev. 12, 914–926 (1998).
Liakopoulos, D., Doenges, G., Matuschewski, K. & Jentsch, S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 17, 2208–2214 (1998).
Tateishi, K., Omata, M., Tanaka, K. & Chiba, T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J. Cell Biol. 155, 571–579 (2001).
Del Pozo, J. C. & Estelle, M. The Arabidopsis cullin AtCUL1 is modified by the ubiquitin-related protein RUB1. Proc. Natl. Acad. Sci. USA 96, 15342–15347 (1999).
Hosp, J. et al. A tobacco homolog of DCN1 is involved in pollen development and embryogenesis. Plant Cell Rep. 33, 1187–1202 (2014).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Michelmore, R. W., Paran, I. & Kesseli, R. V. Identification of markers linked to disease-resistance genes by bulked segregate analysis—a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl Acad. Sci. USA. 88, 9828–9832 (1991).
Liu, S., Yeh, C. T., Tang, H. M., Nettleton, D. & Schnable, P. S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7, e36406 (2012).
Wang, R. et al. Bulk segregate RNA-seq reveals expression and positional candidate genes and allele specific expression for disease resistance against enteric septicemia of catfish. BMC Genomics 14, 929 (2013).
Du, H. et al. Bulked segregant RNA-seq reveals differential expression and SNPs of candidate genes associated with water logging tolerance in maize. Front. Plant Sci. 8, 1022 (2017).
Liu, X. et al. Rapid identification of a candidate nicosulfuron sensitivity gene (Nss) in maize (Zea mays L.) via combining bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 132, 1351–1361 (2019).
Wu, P. et al. Development of molecular markers linked to powdery mildew resistance gene Pm4b by combining SNP discovery from transcriptome sequencing data with bulked segregant analysis (BSR-Seq) in wheat. Front. Plant Sci. 9, 95 (2018).
Huang, Z. et al. Fine mapping of a clubroot resistance gene in Chinese cabbage using SNP markers identified from bulked segregant RNA sequencing. Front. Plant Sci. 8, 1448 (2017).
Kim, S., Kim, C. W., Park, M. & Choi, D. Identification of candidate genes associated with fertility restoration of cytoplasmic male-sterility in onion (Allium cepa L.) using a combination of bulked segregant analysis and RNA-seq. Theor. Appl. Genet. 128, 2289–2299 (2015).
Qiu, Y., Liao, L., Jin, X., Mao, D. & Liu, R. Analysis of the meiotic transcriptome reveals the genes related to the regulation of pollen abortion in cytoplasmic male-sterile pepper (Capsicum annuum L.). Gene 641, 8–17 (2018).
Liu, H. Comparative Proteome and Polyubiquitin Related Proteome Analysis of Normal and Male Sterile Wheat (Triticum aestivum L.). PhD dissertation (Northwest A&F Univ. Yanglin, China, 2014). (in Chinese)
Nakayama, K. I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 6, 369–381 (2006).
Merlet, J., Burger, J., Gomes, J. E. & Pintard, L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol. Life Sci. 66, 1924–1938 (2009).
Xirodimas, D. P. et al. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep 9, 280–286 (2008).
Um, J. W. et al. Neddylation positively regulates the ubiquitin E3 ligase activity of parkin. J. Neurosci. Res. 90, 1030–1042 (2012).
Sui, Y., Liu, Y. & Xu, G. A lysine-to-arginine mutation on NEDD8 markedly reduces the activity of cullin RING E3 ligase through the impairment of neddylation cascades. Biochem. Biophys. Res. Commun. 461, 653e658 (2015).
Hershko, A. & Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998).
Pickart, C. M. & Eddins, M. J. Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695, 55–72 (2004).
Hou, C. C. & Yang, W. New insights to the ubiquitin–proteasome pathway (UPP) mechanism during spermatogenesis. Mol. Biol. Rep. 40, 3213–3230 (2013).
Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21 (1994).
Ciechanover, A. The ubiquitin–proteasome pathway: on protein death and cell life. EMBO J. 17, 7151–7160 (1998).
Chen, H. Y., Sun, J. M., Zhang, Y., Davie, J. R. & Meistrich, M. L. Ubiquitination of histone H3 in elongating spermatids of rat testes. J. Biol. Chem. 273, 13165–13169 (1998).
Khor, B. et al. Proteasome activator PA200 is required for normal spermatogenesis. Mol. Cell. Biol. 26, 2999–3007 (2006).
Boissonnas, C. C., Jouannet, P. & Jammes, H. Epigenetic disorders and male subfertility. Fertil. Steril. 99, 624–631 (2013).
Zhang, J. et al. Correlation between ubiquitination and defects of bull spermatozoa and removal of defective spermatozoa using anti-ubiquitin antibody-coated magnetized beads. Anim. Reprod. Sci. 192, 44–52 (2018).
Schwechheimer, C. NEDD8—its role in the regulation of Cullin-RING ligases. Curr. Opin. Plant Biol. 45, 112–119 (2018).
Brown, J. S. & Jackson, S. P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 5, 150018 (2015).
Li, T., Guan, J., Huang, Z., Hu, X. & Zheng, X. RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J. Cell Sci. 127, 2238–2248 (2014).
Wang, C. & Liu, Z. Arabidopsis ribonucleotide reductases are critical for cell cycle progression, DNA damage repair, and plant development. Plant Cell 18, 350–365 (2006).
Balk, J. & Leaver, C. J. The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome C release. Plant Cell 13, 1803–1818 (2001).
Barchenger, D. W. et al. Genome-wide identification of chile pepper pentatricopeptide repeat domains provides insight into fertility restoration. J. Am. Soc. Hort. Sci. 143, 418–429 (2018).
Jo, Y. D. et al. Fine mapping of Restorer‑of‑fertility in pepper (Capsicum annuum L.) identified a candidate gene encoding a pentatricopeptide repeat (PPR)‑containing protein. Theor. Appl. Genet. 129, 2003–2017 (2016).
Zhang, Z. et al. Fine mapping of the male fertility restoration gene CaRf032 in Capsicum annuum L. Theor. Appl. Genet. 133, 1177–1187 (2020).
Cheng, J. et al. Fine mapping of restorer-of-fertility gene based on high-density genetic mapping and collinearity analysis in pepper (Capsicum annuum L.). Theor. Appl. Genet. 133, 889–902 (2020).
Wei, B. et al. Comparative transcriptional analysis of Capsicum flower buds between a sterile flower pool and a restorer flower provides insight into the regulation of fertility restoration. BMC Genomics 20, 837 (2019).
Hill, J. T. et al. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697 (2013).
Acknowledgements
The authors thank Franchesca Ortega from the chile pepper breeding and genetics program NMSU for editing the manuscript. This research was funded by the Scientific Research Start-up Funds for Openly recruited Doctors of Science and Technology Innovation Funds of Gansu Agricultural University, China (GAU-KYQD-2018-17) and the National Natural Sciences Foundation of China (31560555, 31760572).
Author information
Authors and Affiliations
Contributions
B.W. and J.Y. conceived and designed the work; B.W. performed the work; B.W. wrote the first draft of the manuscript; and P.W.B. and revised the manuscript; Z.Z. and Y.W. performed the KASP assay; G. Z analyzed the data; L.W. provided some plant materials. All authors read, reviewed, and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, B., Bosland, P.W., Zhang, Z. et al. A predicted NEDD8 conjugating enzyme gene identified as a Capsicum candidate Rf gene using bulk segregant RNA sequencing. Hortic Res 7, 210 (2020). https://doi.org/10.1038/s41438-020-00425-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00425-7
This article is cited by
-
Genome-wide identification of RUB activating enzyme and conjugating enzyme gene families and their expression analysis under abiotic stresses in Capsicum annuum
Protoplasma (2023)
-
Fine mapping of Rf2, a minor Restorer-of-fertility (Rf) gene for cytoplasmic male sterility in chili pepper G164 (Capsicum annuum L.)
Theoretical and Applied Genetics (2022)
-
Identification and bioinformatic analysis of the CaCesA/Csls family members and the expression of the CaCslD1 in the flower buds of CMS/Rf system in pepper
Functional & Integrative Genomics (2022)
-
The Unstable Restorer-of-fertility locus in pepper (Capsicum annuum. L) is delimited to a genomic region containing PPR genes
Theoretical and Applied Genetics (2022)
-
Development and application of KASP markers associated with Restorer-of-fertility gene in Capsicum annuum L.
Physiology and Molecular Biology of Plants (2021)
