
Abstract
We report the acquisition of a high-quality haploid chromosome-scale genome assembly for the first time in a tree species, Eucommia ulmoides, which is known for its rubber biosynthesis and medicinal applications. The assembly was obtained by applying PacBio and Hi–C technologies to a haploid that we specifically generated. Compared to the initial genome release, this one has significantly improved assembly quality. The scaffold N50 (53.15 MB) increased 28-fold, and the repetitive sequence content (520 Mb) increased by 158.24 Mb, whereas the number of gaps decreased from 104,772 to 128. A total of 92.87% of the 26,001 predicted protein-coding genes identified with multiple strategies were anchored to the 17 chromosomes. A new whole-genome duplication event was superimposed on the earlier γ paleohexaploidization event, and the expansion of long terminal repeats contributed greatly to the evolution of the genome. The more primitive rubber biosynthesis of this species, as opposed to that in Hevea brasiliensis, relies on the methylerythritol-phosphate pathway rather than the mevalonate pathway to synthesize isoprenyl diphosphate, as the MEP pathway operates predominantly in trans-polyisoprene-containing leaves and central peels. Chlorogenic acid biosynthesis pathway enzymes were preferentially expressed in leaves rather than in bark. This assembly with higher sequence contiguity can foster not only studies on genome structure and evolution, gene mapping, epigenetic analysis and functional genomics but also efforts to improve E. ulmoides for industrial and medical uses through genetic engineering.
Similar content being viewed by others

Introduction
Polyisoprene, a polymer of isoprene (C5H8), is the primary chemical constituent of natural rubber. Based on the chemical structure of the two isoprene isomers, natural rubber can be classified into cis-polyisoprene (CPI) and trans-polyisoprene (TPI)1. Currently, the Brazilian rubber tree is the only commercial source of natural CPI2,3. This tropical tree species faces several challenges, however, including a narrow range of suitable habitats, densely planted areas for latex tapping, and threats from serious diseases such as blight and insect pests4,5. For these reasons, the shortage of natural rubber caused by an increasing global demand has led both governments and industrial enterprises to look for substitutes for this essential material to synthesize various rubber products.
Eucommia ulmoides is a tertiary relic perennial tree of high value. It is an important temperate economic tree species that is capable of producing not only wood but also valuable raw biomaterials for extracting rubber and the active ingredients of a traditional Chinese medicine called Duzhong6,7. Unlike the Brazilian rubber tree, E. ulmoides is a well-known tree species for producing trans-rubber, which has unique characteristics such as high hardness, a low thermal expansion/contraction coefficient, good insulation, and resistance to acid and alkaline conditions8,9,10. For this reason, it can be used to manufacture cable insulation and golf balls11,12. Moreover, since blended rubber of CPI and TPI has some augmented mechanical properties and a different thermal conductivity and diffusivity1 compared to those of rubber with CPI only, TPI has been made into cis-rubber by vulcanization to achieve improved elasticity, resilience, tensile strength, viscosity, hardness and weather resistance13.
As previously reported6, TPI is especially enriched in the leaves, bark, and peels of E. ulmoides trees. For this reason, E. ulmoides is considered an ideal alternative or supplementary tree species for producing the raw materials of natural rubber14,15. Moreover, E. ulmoides can grow in many countries located in the northern temperate zone, where Brazilian rubber trees cannot be domesticated. In addition to its value in producing rubber, E. ulmoides is an important source of some active ingredients in traditional Chinese medicine. Its leaves and bark are especially rich in chlorogenic acid (CGA), an antioxidant that is known to slow the release of glucose into the bloodstream after a meal16 and reduce blood pressure17. In addition, it contains aucubin, rutin, quercetin and other unknown ingredients that may have great value for biopharmaceuticals18,19. Therefore, a high-quality genome is indispensable for genetic improvements related to E. ulmoides rubber productivity and medicinal uses in addition to wood.
In 2018, a draft of the E. ulmoides genome (v1.0) was released15. Due to high heterozygosity (0.8–1%) and a large percentage of repetitive sequences (greater than 60%)15,20, genome assembly from diploid sequences can easily incorporate chromosomal fragments from two homologous chromosomes in alternation and increase errors at the connections of contigs/scaffolds, thereby resulting in inaccurate assemblies with poor adjacency and increased linkage and base-error rates. To improve the quality of the E. ulmoides genome sequence, a haploid E. ulmoides (2n = x = 17) individual was generated through parthenogenesis and used for genome sequencing. The genome (v2.0) was acquired by assembling PacBio long reads and anchoring scaffolds based on information from Hi–C interaction confirmation. Using this high-quality genome, additional transcriptomic analyses of different tissues were undertaken to investigate rubber and CGA biosynthesis pathways in E. ulmoides. The genome sequence and gene annotation that we generated from the haploid will be instrumental for all studies on genome structure and evolution, gene linkage and mapping, epigenetic and chromatin-based analyses, and functional genomics as well as genetic engineering of E. ulmoides for industrial and medical uses.
Results
Induction of haploidy by high-temperature exposure
To induce haploidy through parthenogenesis, female flower buds of E. ulmoides in the developmental stage of embryo sac formation were treated with different combinations of high temperatures and treatment times, and untreated female flower buds were used as controls. A total of 2675 seeds were harvested after the treatments, and 835 plants were eventually obtained after sowing seeds and transplanting seedlings. Using flow cytometry and analysis of anatomical sections of stem tips, three authentic Eucommia haploid plants were obtained exclusively from the treated group (Fig. 1a–d). Of these, one haploid was obtained from continuous treatment at 48 °C for 6 h, while the other two haploid plants were obtained from continuous treatment at 54 °C for 4 h.

a Somatic chromosome number of the haploids (2n = x = 17). b Ploidy levels obtained from 3-week-old first leaf samples from haploid plants by flow cytometric analysis. c Ploidy levels obtained from 3-week-old first leaf samples from a mixture of haploid and diploid plants by flow cytometric analysis. d A haploid plant (left) and diploid plant (right) of E. ulmoides
Genome sequencing and assembly
We conducted de novo sequencing using the PacBio Sequel platform, which yielded a total of 97.26 Gb of raw data, representing a 95× coverage depth of the 1.02 Gb genome as estimated by 17-Kmer evaluation (Table S1 and Figs. S1 and S2). The PacBio long reads were corrected and assembled into 564 contigs using FALCON and Quiver21, with a contig N50 of 13.16 Mb (Table 1 and Table S2), which is 632 times longer than the contig N50 of the v1.0 genome. The total length of these contigs is 947.84 Mb of genome sequence (Tables S2 and S3), accounting for 92.93% of the estimated genome size. In addition, 133.74 Gb of high-quality Hi–C data with ~130× coverage was generated by the Illumina NovaSeq platform (Table S1). Valid high-quality Hi–C data were aligned with PacBio contigs using Burrows-Wheeler Aligner (BWA)22, and duplicates were removed with SAMTOOLS rmdup23 before LACHESIS24 was employed to assemble them into scaffolds based on linkage information and restriction enzyme sites. Finally, 145 long scaffolds were anchored to 17 linkage groups and further formed pseudochromosomes, accounting for 94.94% of the assembly (Table S4). The genome (v2.0) finally acquired contains a total of 501 scaffolds with a scaffold N50 of 53.15 Mb (Table 1 and Table S3), which is 28-fold longer than that in v1.0. Compared to the 104,772 gaps between adjacent scaffolds in v1.0, the number of gaps in v2.0 is only 128. Core Eukaryotic Genes Mapping Approach (CEGMA) evaluation25 was undertaken and showed that the newly assembled genome covered 230 (92.74%) of the 248 core eukaryotic genes (CEGs) completely and 8 (3.23%) of them partially, and the number of undetected CEGs was therefore 10 (4.04%) (Table S5). Genome completeness was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCOs)26, which revealed complete genes (91.3%) and partial genes (1.7%) (Table S6). To further evaluate the assembly schemes, high-quality reads from a small-fragment library (350 bp) were aligned to the assembled genome. The results showed that the alignment rate was 99.18%, and the genome coverage was 99.86% (Table S7). A high degree of consistency between the Hi–C and PacBio results was also illustrated (Fig. S3). These results provide evidence that the new E. ulmoides genome assembled from the haploid is of high quality.
Gene content and annotation
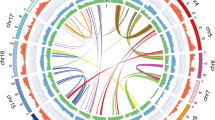
Multiple strategies, including de novo prediction, homology-based methods, and transcript alignment, were employed for gene identification and annotation. Augustus27, GlimmerHMM28 and SNAP (http://homepage.mac.com/iankorf/) software were used for de novo gene prediction, whereas BLAST29 and Genewise30 were utilized to identify homologous open reading frames (Table S8). RNA-seq data derived from 12 different tissue types were assembled with Trinity31, and the assembled sequences were aligned against the E. ulmoides genome by PASA32. A total of 26,001 protein-coding genes, compared to 26,723 in v1.0, were predicted. The average length of the predicted genes was 6,138.21 bp, the average coding sequence (CDS) length was 1,108.80 bp, the average number of exons contained in each gene was 4.85, the average exon length was 228.56 bp, and the average intron length was 1,305.93 bp (Table S9 and Fig. S4). In total, 98.8% of the 26,001 predicted genes had homologs in at least one of the six functional protein databases aligned (Figs. S5, S6 and Table S10), and 24,148 genes could be localized to the chromosome, accounting for approximately 92.87% of the total predicted genes. The GC content, repeat density and gene density for each chromosome were plotted with Circos33 (Fig. 2). Many noncoding RNAs, including 2099 rRNAs, 825 tRNAs, 1032 miRNAs, and 875 snRNAs, were identified and are shown in Table S11.

a Lengths of pseudochromosomes (0.5 Mb window size (WS)); b gene density (0.3 Mb WS); c repeat density (1 Mb WS); d GC content (1 Mb WS). The colored lines in the center show links between syntenic blocks of at least five genes
The use of haploid E. ulmoides and PacBio data greatly facilitated the acquisition of repetitive sequences. A total of 592.43 Mb of repeat sequences, accounting for 62.50% of the genome length, were identified in the E. ulmoides v2.0 genome (Table 1 and Table S12). Combined homology-based and de novo approaches were used to identify transposable elements (TEs); long terminal repeats (LTRs), which are the main TE family in E. ulmoides, had a size of 520 Mb, accounting for 50.33% of the assembled genome. This was much longer than the 361.76 Mb of LTRs accounting for only 30.63% of the v1.0. genome. The copia- and gypsy-like LTRs covered 179.53 Mb and 267.69 Mb, respectively, accounting for 18.94% and 28.24% of the assembled genome. Only 3% of repeats in the v2.0 genome could not be classified into any known category (Table S13). These results indicate that the combination of a haploid genome and PacBio sequencing technology significantly improved the construction of repeat regions. Among the 8,264 intact LTR retrotransposons (LTR-RTs), 5254 and 2980 were gypsy-like and copia-like LTRs, respectively (Table S14). The estimated times for the intact LTR-RT insertion events indicate that the LTRs, copia-like LTRs and gypsy-like LTRs started to expand rapidly ~7.2 million years ago (Mya) and peaked ~5.2, 5.0 and 3.6 Mya, respectively. This time period falls within the Pliocene epoch (5.33–2.58 Mya) (Fig. 3a). The expansion of the copia type occurred before that of the gypsy type, causing intact LTR-RTs of the gypsy type to outnumber those of the copia type (Fig. 3a).

a The insertion times for intact LTR-RTs in the E. ulmoides genome. The insertion times for LTR-RTs were calculated by the formula T = K/2r. T insertion time; r synonymous mutations/site/Mya; K the divergence between the two LTRs. A substitution rate of 8.25 × 10−9 per site per year was used to calculate the insertion times. b Venn diagram of shared orthologs among the five species. Each number represents a gene family number. c Phylogenetic tree of 12 species based on orthologs of single-gene families. The number at the root (28,004) represents the number of gene families related to the common ancestor. The value above each branch denotes the number of gene families gained/lost during each round of genome duplication after diversification from the common ancestor. The red number below each branch denotes the speculated divergence time of each node. Bootstrap values for all nodes are above 50%. d Density distributions of Ks for paralogous genes. The peak values are shown in insets for E. ulmoides and C. canephora. e Density distributions of Ks for paralogous genes. The peak values are shown in insets for E. ulmoides and S. lycopersicum. f Schematic representation of syntenic genes among E. ulmoides, V. vinifera and C. canephora. Gray lines in the background indicate the collinear blocks of at least twenty genes within the E. ulmoides genome and other plant genomes, while the red lines highlight the syntenic gene pairs
Evolution of the E. ulmoides genome
Orthologous clustering of the predicted E. ulmoides proteins with those from 11 other representative species showed that 20,219 of the E. ulmoides protein-coding genes belong to 13,494 gene families with an average of 1.50 genes per family; 538 of these gene families are unique to E. ulmoides (Table S15 and Fig. S7)34. KEGG analysis revealed that E. ulmoides-specific genes are enriched in several pathways, including environmental adaptation, amino acid metabolism, glycan biosynthesis, and lipid metabolism (Table S16). Evolutionary analysis of gene families in E. ulmoides, together with those from 11 other plant species, was undertaken with CAFÉ (v 2.1)35. The results showed that 74 gene families expanded, while 131 gene families contracted (Fig. 3c). Comparative analysis of the protein-coding gene families of E. ulmoides and those in other closely related asterid species, including Catharanthus roseus, Solanum tuberosum, Coffea arabica and Olea europaea, revealed that 8836 of the 20,981 gene families were shared by these five species (Fig. 3b). The families that have expanded significantly are those that are mainly involved in cyanoamino acid metabolism, phenylpropionic acid biosynthesis, starch and sucrose metabolism, and glutathione metabolism (Table S17). This finding suggests that the expanded and unique gene families might be closely associated and have evolved to enhance environmental adaptability and to protect against herbivores and plant pathogens by increasing the production of high secondary metabolite contents.
There has been some controversy regarding classifying E. ulmoides into the lamiid lineage15,36. To determine its evolutionary status, a phylogenetic tree was constructed based on 133 single-copy gene families shared by E. ulmoides and 11 other species. In addition, their divergence time was estimated (Fig. 3c). The results indicate that E. ulmoides and four plant species from different categories of lamiids, including C. roseus, S. tuberosum, C. arabica, and O. europaea, are on the sister branches located in the basal group of the lamiid lineage. This result also supports the recent divergence of the campanulids Daucus carota and Taraxacum kok-saghyz and the recent divergence of Ericales (Actinidia chinensis) and Cornales (Camptotheca acuminata), which are sister groups located at the base of asterids37. The results of the phylogenetic tree are consistent with the systematic classification of the Angiosperm Phylogeny Group (APG IV, 2016). After calibrating the dates with those in the TimeTree database (http://www.timetree.org/), it is estimated that E. ulmoides diverged from campanulids ~100.5 Mya.
Whole-genome duplication (WGD) or polyploidization has occurred in the evolutionary history of most plant species38. The distribution of synonymous substitutions (Ks) and 4-fold synonymous third-codon transversions (4DTv) between duplicated genes was used to extrapolate WGD events. The results show that the E. ulmoides genome has two peaks in the Ks (Fig. 3d, e) and 4DTv distributions (Eulm_Eulm) (Fig. S8a, b), indicating that E. ulmoides may have undergone two WGD events. This is inconsistent with the results for the v1.0 genome15, where the peaks of γ duplication events in the Ks distribution analysis of D. carota, Solanum lycopersicum, and Populus trichocarpa were not conspicuous, presumably because less stringency was used in their analysis. Given that Coffea canephora and Vitis vinifera have only undergone an earlier γ paleohexaploidization event shared by all eudicots, the γ duplication events of E. ulmoides and C. canephora were very close to each other and resembled those in the C. canephora genome39. Collinearity analysis of E. ulmoides, C. canephora and V. vinifera with JCVI40 revealed a 2:1 synteny pattern between either E. ulmoides and C. canephora or E. ulmoides and V. vinifera (Figs. S9 and S10). QUOTAALIGN was then used to draw collinear dot plots to further validate the additional WGD upon γ paleohexaploidization (Figs. S11 and S12). As shown in Table S18, when the ratio of E. ulmoides to either coffee or grape was 1:1, E. ulmoides coverage was only 65.6% and 61.5% compared to 92.5% and 94%, respectively, in C. canephora and V. vinifera. When the ratios of E. ulmoides to C. canephora and V. vinifera were both 2:1, E. ulmoides coverage increased to 96.2% and 97%, respectively; thereafter, the coverage of E. ulmoides remained unchanged (<±0.5%), even when the ratio increased to a high value, such as 8:1. All of these results support that E. ulmoides underwent another WGD event after the earlier γ paleohexaploidization event shared by all eudicots (Fig. 3d–f and Table S18). Based on the repeated gene pairs in the genome, these two WGDs of E. ulmoides occurred 69.7 Mya and between 109.1 and 121.1 Mya, respectively (Table S19).
Metabolic gene clusters and rubber biosynthesis pathways
Plant genes involved in secondary metabolic pathways are sometimes concentrated in specific genomic regions, resulting in biosynthesis gene clusters (BGCs)35,41,42,43. Using plantiSMASH44, we identified 26 BGCs that are associated with various secondary metabolic pathways in E. ulmoides (Table S20 and File S1). Among these 26 BGCs are 9 saccharide-related BGCs, 6 terpene-related BGCs, 4 alkaloid-related BGCs, 1 polyketide-alkaloid-related BGC, 1 saccharide-terpene-related BGC, and 1 lignin-polyketide-related BGC. The other four putative BGCs could not be assigned to a specific secondary metabolic pathway. The sizes of the BGCs identified vary from 32.01 to 2191.17 kb, which usually contain 3–5 core protein domains related to secondary metabolism. These tightly linked BCGs may facilitate cosegregation of coadaptive variation and limit unfit recombinant BCGs, resulting in more stable secondary metabolism.
Trans-polyisoprene (TPI) is known to be synthesized from isoprenyl diphosphate (IPP)45,46 through two pathways, namely, the mevalonate (MVA) pathway in the cytoplasm and the methylerythritol-phosphate (MEP) pathway in plastids47,48. A total of 47 candidate genes involved in the TPI biosynthesis pathway were identified in the genome (v2.0), including 13 genes involved in 6 reactions of the MVA pathway; 11 genes involved in 7 reactions of the MEP pathway; 12 genes involved in initial reactions for producing initiators or precursors, which include geranyl diphosphate synthases (GPSs), geranylgeranyl diphosphate synthases (GGPSs) and farnesyl diphosphate synthases (FPSs)); and 11 genes for ‘rubber elongation’ on TPI particles (Table S21 and Fig. S13).
Earlier studies showed that in addition to xylem and seeds, E. ulmoides leaves and peels contain a certain proportion of high-molecular-weight TPI45. The TPI content is rich in the central peels but relatively poor in the outer peel49,50. Differential expression analysis of genes in different tissues revealed that for the MEP pathway in E. ulmoides, at least one and sometimes two genes that encode enzymes to catalyze each step showed predominant expression in both TPI-containing leaves and central peels, indicating that the synthesis of IPP via the MEP pathway was in operation in both leaves and central peels. Although some MVA pathway genes were also expressed at relatively high levels, there was a lack of TPI-containing tissues in which all genes along the pathway were consistently “switched on”. Overall, MEP pathway genes had the highest or moderate to high expression levels, whereas MVA pathway genes had low to moderate expression levels (Table S21 and Fig. 4 and Fig. S1). It should be noted that prior to this study, we conducted a pilot experiment to examine the differential expression of pathway genes. Differential expression patterns in the pilot experiment were largely the same as those observed in the present study (see Table S21 and Fig. S14). These results suggest that IPP may be primarily and stably synthesized through MEP in the E. ulmoides trees used in our study.

The expression level is presented by log2-transformed fragments mapped per kilobase of transcript length per million total mapped reads (log2-FPKM). ACAT acetyl-coenzyme A (CoA) C-acetyltransferase; HMGS hydroxymethylglutaryl-CoA synthase; HMGR hydroxymethylglutaryl-CoA reductase; MVK mevalonate kinase; PMK 5-phosphomevalonate kinase; MPD mevalonate pyrophosphate decarboxylase; DXS 1-deoxy-d-xylulose 5-phosphate synthase; DXR 1-deoxy-d-xylulose 5-phosphate reductoisomerase; MCT 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase; CMK 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol kinase; MDS 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase; HDS 4-hydroxy-3-methylbut-2-enyl diphosphate synthase; HDR 4-hydroxy-3-methylbut-2-enyl diphosphate reductase; IDI isopentenyl diphosphate isomerase; GPS geranyl diphosphate synthase; FPS farnesyl diphosphate synthase; GGPS, geranylgeranyl diphosphate synthase; SRPP small rubber particle protein; Acetyl-CoA acetyl coenzyme-A; Acetoacetyl-CoA, 3-acetoacetyl-CoA; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; MVA mevalonate; MVA-5P mevalonate-5-phosphate; MVA-5PP mevalonate-5-diphosphate; GA-3-P, glyceraldehyde 3-phosphate; DXP, 1-deoxy-d-xylulose 5-phosphate; MEP 2-C-methyl-d-erythritol 4-phosphate; CME 4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol; PCME 2-phospho-4-(cytidine 5′-diphospho)-2-C-methyl-d-erythritol; CMEC 2-C-methyl-d-erythritol 2,4-cyclodiphosphate; HMED 4-hydroxy-3-methylbut-2-enyl diphosphate; IPP isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GPP geranyl diphosphate; FPP farnesyl diphosphate; GGPP, geranylgeranyl diphosphate. LF leaf; CP central peel; PE peel edge; XM xylem; SD seed
All candidate genes involved in trans-IPP synthesis were obtained through homologous gene comparison, and the results are shown in Table S21. Three GPS, 7 GGPS and 4 FPS genes were identified in this E. ulmoides genome. Previous studies have shown that EuFPS homologs can be divided into two groups, namely, FPS 1/3/5 and FPS 2/4, which may play different roles in TPI synthesis46. FPS 1/3/5 homologs are involved in long-chain TPI biosynthesis, and FPS 2/4 homologs are in the same group as HbFPS1-315. In the newly assembled genome, the evolutionary analysis also indicated that the FPS gene family can be divided into two groups (Figs. S15 and S16), and the expression of FPS1 is more than 200 times higher in leaves and peels with TPI than in xylem and seeds without TPI. This indicates that FPS1 may be a rate-limiting enzyme or at least an important enzyme in TPI synthesis.
As reported earlier, rubber elongation factor (REF)/small rubber particle protein (SRPP) family proteins are critically important for the biogenesis and stability of rubber particles51,52, and there was a positive correlation between REF1 expression and rubber yield in H. brasiliensis53,54. In the E. ulmoides genome (v2.0), as many as nine EuSRPP genes, which is fewer than the 12 H. brasiliensis SRPPs (HbSRPPs), were identified. However, no HbREF1 (138 amino acid) homologs were identified in E. ulmoides, indicating that there are differences in the mechanism of rubber biosynthesis between these two species. Based on the phylogenetic tree (Fig. S17), the SRPP family members of E. ulmoides are divided into two clades. The fragments mapped per kilobase of transcript length per million total mapped reads (FPKM) values of SRPP 1 and SRPP 5 in leaves and peels were significantly higher than 300, but there was no significant difference in the expression levels of SRPP5 between TPI tissue and non-TPI tissue. In contrast, the expression levels of SRPP 1 in TPI tissues (leaves and peels) were 7 times higher than those in non-TPI tissues (xylem and seeds). Presumably, SRPP 1 protein, similar to FPS1, may play a more important role in TPI synthesis. Based on the phylogenetic tree of REF/SRPP proteins from rubber-producing and non-rubber-producing plants5,15,52,55, REF/SRPP genes are scattered among some evolutionary branches of non-rubber-producing O. europaea, whereas most REF/SRPP genes in T. kok-saghyz, H. brasiliensis and E. ulmoides are distributed on one major branch (Fig. S18). This supports the rubber biosynthesis REF/SRPP gene families originating from multiple sources or from different evolutionary events, as demonstrated in previous studies5,15,52.
It is particularly noteworthy that genes within BGCs are involved in the synthesis of rubber initiators. Among the 6 predicted potential terpene secondary metabolite biosynthesis gene clusters, the core of Cluster 15 contains polyisoprene synthase. This gene cluster is 72.5 kb in size and is located at 8,851,940–9,257,031 bp on chromosome 4. The 16 genes in this gene cluster were functionally annotated (Table S22). Five core protein domains were predicted to be related to secondary metabolism by plantiSMASH34, including two encoding polyisoprene synthase (evm.model.Chr4.574 (GPS3) and evm.model.Chr4.575 (GGPS7)), one encoding cytochrome P450 (evm.model.Chr4.579) and two encoding dioxygenase (evm.model.Chr4.588 and evm.model.Chr4.593). In addition, nine members of the SRPP gene family were also present in two locally repeated gene clusters (BGCs): one was composed of three genes (evm.model.000124 f.13, evm.model.000124 f.14 and evm.model.000124 f.15), and the other was composed of four genes (evm.model.chr9.92, evm.model.chr9.94, evm.model.chr9.95, and evm.model.chr9.96), which originated from WGD events or tandem replication. The two remaining members are dispersed repeats on different chromosomes.
Identification of genes involved in CGA synthesis
As a traditional herbal medication, E. ulmoides accumulates CGA, the main phenolic compound, in its leaves and bark56. CGAs have long been known to be antioxidants that slow the release of glucose into the bloodstream after a meal16 and reduce blood pressure17. In addition, CGAs have antibacterial, antiviral and antitumor activities57,58. The biosynthesis of CGAs in E. ulmoides occurs through the phenylpropanoid pathway, where the initial reactions are catalyzed by phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase (C4H), and 4-coumarin-CoA ligase (4CL) to generate p-coumarin-CoA (Fig. 5), which is a precursor for synthesizing CGAs through two different pathways. The first pathway produces p-coumaroyl quinic acid, which is then converted to CGAs. These two reactions are catalyzed by hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT) and p-coumaroyl ester 3′-hydroxylase (C3′H)59, respectively. The second pathway is p-coumarin-CoA → p-coumaric shikimic acid → caffeoyl shikimic acid → caffeoyl CoA → CGAs, and the four reactions are catalyzed by HCT, C3′H, HCT and hydroxycinnamoyl-CoA quinate HCT (HQT), respectively60. Through homologous gene comparison, 23 candidate genes related to 6 key enzyme gene families involved in the CGA biosynthesis pathway were identified in the E. ulmoides genome, including 7 PAL genes, 8 4CL genes, 2 C4H genes, 2 C3′H genes, 1 HCT gene, and 3 HQT genes (Fig. 5 and Table S23). Gene expression profile analysis showed that the expression levels of PAL (PAL2 and PAL5), C3′H2, C4H6, HQT2 and HCT were significantly higher than the expression levels of other genes. These genes may be key genes whose proteins catalyze CGA biosynthesis in E. ulmoides. In addition, some transcription factors (TFs) have also been reported to regulate CGA biosynthesis, including the MYB, ZIP, and WRKY families61,62,63,64. We identified 1,406 TF-encoding genes in E. ulmoides, representing 5.41% of the total genes. The annotation of these TFs is provided in Table S24. We specifically compiled the TFs that control biosynthesis of secondary metabolites (Table S25), which are composed primarily of phenolics, terpenoids, glucosinolates, and alkaloids and have been reported to serve the following roles: (1) protect plants against herbivores and pathogens65,66 and (2) constitute effective compounds in herbs in traditional medicine67. This information provides valuable information for further research on secondary metabolites in E. ulmoides.

The expression level is presented as log2-transformed fragments mapped per kilobase of transcript length per million total mapped reads (log2-FPKM). L1-L3 leaf; B1-3 bark. The enzymes involved are as follows: PAL phenylalanine ammonia-lyase; C4H cinnamate 4-hydroxylase; 4CL 4-coumaroyl-CoA ligase; HCT hydroxycinnamoyl-CoA: shikimate hydroxycinnamoyl transferase; C3′H p-coumaroyl ester 3′-hydroxylase; HQT hydroxycinnamoyl-CoA:quinate hydroxycinnamoyl transferase
Discussion
Haploids play an important role in plant breeding and genome sequencing research. Typically, in vitro anther culture is the most widely used method to induce haploids68. However, anther culture cannot be achieved for the many plant species lacking an established tissue culture system. In this study, an in vivo branch bud heat treatment device69 was used for high-temperature treatment of E. ulmoides during the developmental stage of female embryo bud sacs, and three haploid lines were obtained. This shows that high-temperature treatment during embryo sac development is suitable and effective for inducing haploids in E. ulmoides. It also provides a reference for other plant species that do not have an established tissue culture system to induce apomixis at high temperature.
Various types of genetic and genomic research necessitate an accurate de novo whole-genome assembly as a foundation for developing genetic resources to understand evolution, genetics, inheritance, genomics, epigenetics and functional genomics. However, acquisition of a high-quality genome assembly is an arduous task, especially for species with high heterozygosity (or polymorphism) and/or highly repetitive sequences, including gene duplications, transposons, and short sequence repeats70,71. To circumvent these challenges, we specifically developed haploid E. ulmoides plants through parthenogenesis as described above. We used plant material from one of these haploids for genome sequencing, which can enhance the quality of genome assembly and the accuracy of gene annotation owing to the lack of heterozygosity. In addition, the use of advanced PacBio long reads and Hi–C data further enhances the quality of the genome. Compared to the initial genome released (v1.0)15, the current version (v2.0) has significantly improved assembly quality. For example, the scaffold number was reduced from 29,152 to 501, the scaffold N50 (53.15 MB) increased 28-fold compared to that of v1.0, and the number of gaps between scaffolds decreased from 104,772 in v1.0 to 128 in v2.0 (Table S26). A previous study in fish showed that the use of a haploid sequence for whole-genome assembly can increase the scaffold N50 by 3- to 10-fold71. We thus assume that the 28-fold gain in the scaffold N50 may also be ascribed to the other technologies we used, such as Hi–C. In addition, we obtained 520 Mb of repetitive sequence, which is 158.24 Mb longer than that of the v1.0 genome. Further evaluation with CEGMA revealed 238 (95.97%, complete + partial) and 230 (92.74%, complete) of 248 core eukaryotic genes in genome v2.0, in contrast with 232 (93.55%, complete + partial) and 196 (79.03%, complete) in genome v1.0 (Table S5). BUSCO analysis identified 93% of the 1,440 single-copy genes in genome v2.0, of which 91.3% were complete copies, which was higher than the 92.8% single-copy genes and 90.0% complete copies identified in genome v1.0 (Table S6). Although the 26,001 predicted genes in genome v2.0 is lower than the 26,723 predicted in genome v1.0, the results of the BUSCO and CEGMA evaluations indicate that the v2.0 genome annotation is more accurate and complete.
The E. ulmoides genome contains a high percentage of repetitive sequences, which poses a great challenge to genome assembly when short reads are used. This is because many short-read assemblers employ an algorithm called de-Bruijn graph representation, which connects short sequence fragments based on their overlapping subsequences. When the de-Bruijn graph is used to extend the assembly, the assembler must deal with repetitive sequences by finding the optimal path from multiple choices71. However, this is often impossible, especially when extending contigs through repetitive sequences longer than the read length, resulting in many fragmented assemblies that end in unresolved repetitive sequences72. Therefore, the use of haploid PacBio long reads can significantly reduce the complexity and enhance walk-throughs of repetitive sequences. The majority of repetitive sequences are present in the genome as TEs, which play important roles in genome reorganization and evolution because TE insertions can change genome structure and generate mutations from which new genes can be derived73. In addition, TEs can modify gene regulatory networks by dispersing vast numbers of promoter and enhancer transcription factor binding sites, insulator sequences, and repressive elements74. Given its important roles, a genome assembly without accurately assembled repetitive sequences will be deficient for studying genome evolution and gene regulation. The total length of LTRs in the v2.0 genome is ~520 Mb, which greatly exceeds the 361.76 Mb in genome v1.0. The v2.0 genome contains only 3% noncategorical repeats, which is far less than the 17.16% in the v1.0 genome. Further analysis shows that most LTR-RT expansions occurred in the Pliocene 5.2 Mya. From the perspective of fossil history, the distribution range of E. ulmoides was almost throughout the Northern Hemisphere during the Miocene, and it began to shrink in the Neogene75. The reason is that during the Pliocene, the temperature and atmospheric carbon dioxide concentration continued to decline, and in the Pleistocene, Earth entered the glacial period, which changed plant growth and distribution zones76. Changes in environmental pressures led to the activation of TEs and plant genome reorganization as well as gene expansion77,78. Half of the resistance gene analogs in this genome were derived from TE insertions (File S2), suggesting that TE burst might have played an important role in E. ulmoides evolution and stress response.
The evolutionary history of E. ulmoides is further confirmed to be different from that of genome v1.015. According to the taxonomy of the Angiosperm Phylogeny Group (APG IV, 2016), E. ulmoides is a latex-producing deciduous species in Garryales, which belongs to lamiids. Wuyun et al.15 concluded that E. ulmoides is a sister taxon of lamiids and campanulids. In this study, a phylogenetic tree based on genome v2.0 showed that E. ulmoides belongs to lamiids, which is consistent with the systematic classification of angiosperms by The Angiosperm Phylogeny Group APG IV (2016). The inconsistency might arise from the improved genome (2.0) and the selection of more lamiid plants to build phylogenetic trees. WGD or polyploidization has played an important role in plant genome evolution79. A previous study on genome v1.0 concluded that there was no additional WGD upon the γ paleohexaploidization event15. However, the present study provides multiple lines of new evidence that the E. ulmoides genome was marked by a lineage-specific paleotetraploidization event superimposed on the earlier paleohexaploidization event shared by all eudicots. By comparing syntenic patterns between E. ulmoides and grape/coffee, we obtained a stable 2:1 synteny ratio (Fig. 3f and Table S18). These results provide a basis for future comparative genomics analyses of asterid evolution.
The IPP derived from the MVA and MEP pathways is considered a substrate for synthetic TPI45,80. 13C isotopic labeling showed that TPI was obtained from both the MVA and MEP pathways in E. ulmoides45. Studies in H. brasiliensis and dandelion-type plants have shown that the MVA pathway is mainly involved in the synthesis of cis-rubbers5,52,80,81. Previous research using the v1.0 genome showed that Eucommia is similar to H. brasiliensis and T. kok-saghyz, and the main route of IPP synthesis is the MVA pathway15. However, a different result was obtained in this study. At least one enzyme in each process of the MEP pathway in E. ulmoides was predominantly expressed in the TPI-containing leaves and central peel. Therefore, the MEP pathway, rather than the MVA pathway, plays a major role in the synthesis of IPP for TPI biosynthesis. Pilot studies conducted previously produced similar results (Table S21 and Fig. S14). Such a discrepancy from the results of Wuyun et al may result from variation in the tree population. It is possible that the trees we studied are from different provenances and have adapted to different ecotypes or climate types. This variation may provide an opportunity for selective breeding for the improvement of TPI synthesis.
To date, four protein families related to long-chain CPI rubber elongation have been characterized: cis-prenyltransferases (CPTs), SRPP, REF and Nogo-B receptor (NgBR)-like protein51,53,82,83. CPT is responsible for adding IPP to the substrate in the cis-configuration to form long-chain CPI82,84. REF/SRPP is involved in the stabilization of the rubber particle membrane85,86. NgBR-like protein is a homolog of the human Nogo-B receptor and has been demonstrated to be essential for rubber biosynthesis in dandelion83. The NgBR-like protein functions as a mediating protein between REF and CPT to form the ternary complex to increase CPT activity87. However, the details of long-chain TPI biosynthesis after IPP formation in E. ulmoides via the MVA and MEP pathways remain largely unknown. Wuyun et al.15 found that some key amino acids in the conserved features/sites of FPS 1, 3, and 5 differed from those of FPS 2 and 4 and all three HbFPS1-3. At the same time, long-chain TPI was obtained after overexpression of FPS5 in tobacco, indicating that FPS 1, 3, and 5 may participate in the synthesis of long-chain TPI. Wuyun et al.15 also found that the expression levels of EuSRPP1, 2, and 7 instead of REFs corresponded to the accumulation of Eu-rubber. The expression levels of three REF genes were almost all zero in expression in all tissues examined15. In our study, we failed to annotate FPS5 in the first round of gene discovery because of its low expression levels in most tissues (Table S21), indicating the possibility of increasing TPI synthesis by overexpressing FPS1. In the v2.0 genome, only 9 SRPP gene family members were detected, and no genes that are homologous to HbREF1 (138 amino acids) were identified, indicating that they may be pseudogenes with incomplete gene sequences arising from the fragmentation of the contig/scaffold sequences in the v1.0 genome. In addition, 3 CPT genes and an NgBR homologous gene were found in the v2.0 genome, and their expression levels were much lower than those of FPS1 and SRPP1 (Table S21). This suggests that although CPT, REF, and NgBR-like proteins were reported to participate in long-chain CPI biosynthesis, whether they are functionally involved in the biosynthesis of long-chain TPI is unclear and needs to be further investigated. Given that the rubber in E. ulmoides is pure TPI6,88, it is still not possible to sketch the biochemical scenario for long-chain TPI synthesis in E. ulmoides with only the genes whose counterparts have been reported in other species as being involved in CPI synthesis. Clearly, more functional genes and enzymes need to be identified and characterized to delineate the underlying biochemical reactions and pathways of TPI synthesis. Corresponding to CPT, there should be at least one trans-prenyltransferase (TPT) enzyme that can add IPP in a trans configuration during the synthesis of long-chain TPI in E. ulmoides. TPT is a large gene family. Its main function is to add IPP to the precursor in the trans configuration. According to the chain length of the product, it can be divided into short-chain and long-chain TPTs, where short-chain TPTs are widely found in plants, mainly including geranyl GPS, FPS, and GGPS5,52,89,90. However, the long-chain TPT responsible for adding IPP to precursors for long-chain TPI rubber elongation is still unknown. Therefore, in addition to identifying long-chain TPTs, the functions of FPS and SRPP in Eu-rubber synthesis also need to be further characterized because existing evidence indicates that they may play a role during the synthesis of long-chain TPI15. Such divergence among species implies that the biosynthesis of polyisoprene in angiosperms is polyphyletic in origin.
In conclusion, the haploid E. ulmoides genome sequencing project has provided a highly accurate and contiguous reference genome sequence for evolutionary and genomic studies. Our preliminary analyses have added valuable information to the genomic resources of lamiids and led to novel conclusions regarding its evolution in terms of WGD, BGCs and rubber synthesis pathways. We believe that this work will be instrumental not only for accelerating evolutionary, genetic and genomic studies in E. ulmoides but also for advancing our understanding of the molecular mechanisms underlying the biosynthesis and regulation of rubber and chlorogenic acid as well as other valuable secondary metabolites such as rutin and quercetin.
Materials and methods
Induction of haploid by high-temperature exposure
The female flower buds of E. ulmoides in the developmental stage of embryo sac formation were treated at either 48 °C or 54 °C for 4–6 h, and untreated female flower buds were used as controls. Seeds were harvested and planted in a room at 25 °C for germination. Karyotyping was conducted with the aid of flow cytometry and anatomical sections to identify authentic Eucommia haploid plants.
Plant material
Fresh leaves of haploid E. ulmoides plants originally induced by parthenogenesis were grown in a greenhouse for genome sequencing and Hi–C analysis. Apical buds, young leaves, mature leaves, old leaves, bark, roots, peels, seeds, lateral buds, male flowers (SRX2447963, SRX2447934, SRX2447933, and SRX1160206), female flowers (SRX1160207), and stem tissue of E. ulmoides were harvested, immediately frozen in liquid nitrogen and stored at −80 °C for RNA-seq experiments. The resulting RNA-seq data were deposited into the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) public repository.
SMRT long-read sequencing
The DNeasy Plant Mini Kit (TIANGEN, Beijing, China) was used to extract high-quality genomic DNA from young leaves of haploid E. ulmoides. For PacBio sequencing, a single-molecule real-time (SMRT) bell library with a 40-kb insert fragment length was constructed according to the manufacturer’s protocol (PacBio, CA).
Illumina short-read sequencing
For Illumina (San Diego, CA) sequencing, 350-bp libraries with an average insert length of 350 bp were prepared using the NEBNext Ultra DNA Library Prep Kit and sequenced using the Illumina HiSeq X Ten platform at the Novogene Bioinformatics Institute, Beijing.
Hi–C library construction and sequencing
A Hi–C library was prepared using the DovetailTM Hi–C Library Preparation Kit. Nuclear chromatin was fixed with formaldehyde. Fixed chromatin was digested with DpnII, and the sticky ends were filled with biotinylated nucleotides at the digested DNA ends prior to ligation. The chromatin was then reverse-crosslinked to free DNA from proteins, and the purified DNA was processed to remove any free biotin from the linked fragments. The DNA was then cut into 350-bp fragments, and the biotinylated fragments were enriched by pulling down with streptavidin-coated magnetic beads, followed by PCR amplification to generate a library. The size of the inserted fragment was checked using a Qubit 2.0 fluorometer and an Agilent 2100 fluorometer. The effective concentrations of the libraries were accurately quantified by Q-PCR to confirm their quality, and the libraries were sequenced on the Illumina NovaSeq platform.
Genome assembly
Before assembling, the ‘daligner’ option in the FALCON assembler was used to correct the errors in PacBio long reads using PacBio short reads less than 6000 bp in length to generate consensus sequences91. Then, FALCON was used to identify the overlaps between all pairs of preassembled error-corrected reads. The read overlaps were used to construct a directed string graph following Myers’ algorithm. Contigs were constructed by finding the paths from the string graph (falcon_sense_option = - output_multi -min_idt 0.70 -min_cov 4 -max_n_read 300 -n_core 12; overlap_filtering_setting = -max_diff 100 -max_cov 100 -min_cov 4 -n_core 12). Error correction of the preceding assembly was performed by the consensus–calling algorithm Quiver with PacBio reads21. The Illumina reads were also used to correct the contigs with Pilon92. The heterozygosity in the error-corrected contigs was then removed by purge_haplotigs with Illumina clean reads.
The Hi–C clean data were aligned to the preceding assembly using BWA software22. SAMTOOLS was used to remove duplicates (parameters: rmdup) and non-aligned data23, and only the read pairs with both reads in the pair aligned to contigs were considered for scaffolding. The physical coverage of each read pair was defined as the total bases spanned by the sequences of the reads and the gap between the two reads when mapped to contigs. The per base physical coverage of each base in the contig was defined as the number of read pairs that physically included it. Given this, misassembly was detected by a sudden drop in the per-base physical coverage in a contig. According to the physical coverage of the alignment result, misassemblies were sheared to correct errors by SALSA93. Based on the linkage information and restriction enzyme sites, the string graph formulation was used to construct the scaffold graph with LACHESIS24. Finally, the scaffolds were anchored to 17 chromosomes, and then the interaction matrix heat map of all chromosomes was visualized with a resolution of 500 kb.
Evaluation of the genome assembly
To evaluate the accuracy and completeness of the assembled Eucommia genome, Burrows-Wheeler Aligner (BWA)22 software was used to compare a small-fragment library with an average insert length of 350 bp to the assembled genome. The alignment rate, extent of genome coverage, and depth distribution were used to assess assembly integrity and sequencing uniformity. CEGMA25 and BUSCO26 were used to assess the integrity of the final genome assembly.
Additional detailed methods, including annotation of repetitive sequences, gene prediction, phylogenetic tree reconstruction, species divergence time estimation, expansion and contraction of gene families, genome synteny and WGD assessment, transcriptome sequencing, and analysis, identification of resistance genes, identification of CGA synthesis-related genes, identification of transcription factors and phylogenetic analysis, are available in File S2.
Data availability
All sequencing reads and the genome assembly have been deposited in the NCBI database under accession number PRJNA599775.
References
Baboo, M., Dixit, M., Sharma, K. & Saxena, N. S. Mechanical and thermal characterization of cis-polyisoprene and trans-polyisoprene blends. Polym. Bull. 66, 661–672 (2011).
Mooibroek, H. & Cornish, K. Alternative sources of natural rubber. Appl. Microbiol. Biotechnol. 53, 355–365 (2000).
Van Beilen, J. B. & Poirier, Y. Establishment of new crops for the production of natural rubber. Trends Biotechnol. 25, 522–529 (2007).
Lieberei, R. South American leaf blight of the rubber tree (Hevea spp.): new steps in plant domestication using physiological features and molecular markers. Ann. Bot. 100, 1125–1142 (2007).
Tang, C. et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants 2, 16073 (2016).
Bamba, T., Fukusaki, E., Nakazawa, Y. & Kobayashi, A. In-situ chemical analyses of trans-polyisoprene by histochemical staining and Fourier transform infrared microspectroscopy in a rubber-producing plant, Eucommia ulmoides Oliver. Planta 215, 934–939 (2002).
Du, H. Y., Hu, W. Z. & Yu, R. The Report on Development of China’s Eucommia Rubber Resources and Industry (2014–2015) (Social Sciences Academic Press, 2015).
Kent, E. G. & Swinney, F. B. Properties and applications of trans-1,4-polyisoprene. Ind. Eng. Chem. Prod. Res. Dev. 5, 134–138 (1966).
Enoki, M., Doi, Y. & Iwata, T. Oxidative degradation of cis- and trans-1,4-polyisoprenes and vulcanized natural rubber with enzyme-mediator systems. Biomacromolecules 4, 314–320 (2003).
Rose, K. & Steinbuchel, A. Biodegradation of natural rubber and related compounds: recent insights into a hardly understood catabolic capability of microorganisms. Appl. Environ. Microbiol. 71, 2803–2812 (2005).
Schlesinger, W. & Leeper, H. M. Chicle - cis- and trans-polyisoprenes from a single plant species. Ind. Eng. Chem. 43, 398–403 (1951).
Roth, W. B., Carr, M. E., Davis, E. A. & Bagby, M. O. New sources of gutta-percha in Garrya flavescens and G. wrightii. Phytochemistry 24, 183–184 (1985).
Yan, R. F. Prospects and research progress on Eucommia ulmoides gum. Prog. Chem. 7, 65–71 (1995).
Nakazawa, Y. et al. Production of Eucommia-rubber from Eucommia ulmoides Oliv (Hardy Rubber Tree). Plant Biotechnol. 26, 71–79 (2009).
Wuyun, T. N. et al. The hardy rubber tree genome provides insights into the evolution of polyisoprene biosynthesis. Mol. Plant 11, 429–442 (2018).
Ong, K. W., Hsu, A. & Tan, B. K. Chlorogenic acid stimulates glucose transport in skeletal muscle via AMPK activation: a contributor to the beneficial effects of coffee on diabetes. PLoS ONE 7, e32718 (2012).
Onakpoya, I. J., Spencer, E. A., Thompson, M. J. & Heneghan, C. J. The effect of chlorogenic acid on blood pressure: a systematic review and meta-analysis of randomized clinical trials. J. Hum. Hypertens. 29, 77–81 (2015).
Li, F. D. & Du, H. Y. Eucommia ulmoides (China Press of Traditional Chinese Medicine, 2001).
Kwan, C. Y., Chen, C. X., Deyama, T. & Nishibe, S. Endothelium-dependent vasorelaxant effects of the aqueous extracts of the Eucommia ulmoides Oliv. leaf and bark: implications on their antihypertensive action. Vasc. Pharmacol. 40, 229–235 (2003).
Wuyun, T. N., Du, H. Y., Liu, H. M., Wang, L. & Bao, W. Q. Important Research Progress in Genome Sequencing of Eucommia ulmoides (2014-2015) (Social Sciences Academic Press, 2015).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Keller, O., Kollmar, M., Stanke, M. & Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763 (2011).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Liu, Z. H. Floral Organogenesis and Phylogenetic Analysis in Eucommia ulmoides Oilv (Peking University, 2010).
Zhang, N., Zeng, L., Shan, H. & Ma, H. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. N. Phytol. 195, 923–937 (2012).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Iorizzo, M. et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 48, 657–666 (2016).
Tang, H., Krishnakumar, V. & Li, J. jcvi: JCVI utility libraries (Version v0.5.7). Zenodo (2015).
Chae, L., Kim, T., Nilo-Poyanco, R. & Rhee, S. Y. Genomic signatures of specialized metabolism in plants. Science 344, 510–513 (2014).
Nutzmann, H. W., Huang, A. & Osbourn, A. Plant metabolic clusters—from genetics to genomics. N. Phytol. 211, 771–789 (2016).
Kautsar, S. A., Suarez Duran, H. G., Blin, K., Osbourn, A. & Medema, M. H. plantiSMASH: automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 45, W55–W63 (2017). Nucleic Acids Research.
Nutzmann, H. W. & Osbourn, A. Gene clustering in plant specialized metabolism. Curr. Opin. Biotechnol. 26, 91–99 (2014).
Bamba, T. et al. Contribution of mevalonate and methylerythritol phosphate pathways to polyisoprenoid biosynthesis in the rubber-producing plant Eucommia ulmoides Oliver. Z. Naturforsch. C. J. Biosci. 65, 363–372 (2010).
Suzuki, N. et al. Construction and analysis of EST libraries of the trans-polyisoprene producing plant, Eucommia ulmoides Oliver. Planta 236, 1405–1417 (2012).
Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 16, 565–574 (1999).
Kuzuyama, T. Mevalonate and nonmevalonate pathways for the biosynthesis of isoprene units. Biosci. Biotechnol. Biochem. 66, 1619–1627 (2002).
Cui, Y., Wang, M. & Sun, K. Morphological study of gutta containing cells in Eucommia ulmoides Oliv. Chin. Bull. Bot. 16, 439–443 (1999).
Du, H. Y., Du, L. Y. & Li, F. D. Dynamic of gutta-percha formation and accumulation in samara of Eucommia ulmoides. For. Res. 17, 185–191 (2004).
Oh, S. K. et al. Isolation, characterization, and functional analysis of a novel cDNA clone encoding a small rubber particle protein from Hevea brasiliensis. J. Biol. Chem. 274, 17132–17138 (1999).
Lin, T. et al. Genome analysis of Taraxacum kok-saghyz Rodin provides new insights into rubber biosynthesis. Nat. Sci. Rev. 5, 78–87 (2017).
Dennis, M. S. & Light, D. R. Rubber elongation factor from Hevea brasiliensis. Identification, characterization, and role in rubber biosynthesis. J. Biol. Chem. 264, 18608–18617 (1989).
Priya, P., Venkatachalam, P. & Thulaseedharan, A. Differential expression pattern of rubber elongation factor (REF) mRNA transcripts from high and low yielding clones of rubber tree (Hevea brasiliensis Muell. Arg.). Plant Cell Rep. 26, 1833–1838 (2007).
Unver, T. et al. Genome of wild Olive and the evolution of oil biosynthesis. Proc. Natl Acad. Sci. USA 114, E9413–E9422 (2017).
Wu, L. Q., Zhu, W. X., Zhang, Y. X., Fu, D. D. & Zhang, Z. X. Analysis of chlorogenic acid content and extraction detection method in Eucommia ulmoides Oliv. Food Sci. 26, 187–192 (2005).
Huang, M. T., Smart, R. C., Wong, C. Q. & Conney, A. H. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 48, 5941–5946 (1988).
Tajik, N., Tajik, M., Mack, I. & Enck, P. The potential effects of chlorogenic acid, the main phenolic components in coffee, on health: a comprehensive review of the literature. Eur. J. Nutr. 56, 2215–2244 (2017).
Franke, R. et al. The Arabidopsis REF8 gene encodes the 3-hydroxylase of phenylpropanoid metabolism. Plant J. 30, 33–45 (2002).
Niggeweg, R., Michael, A. J. & Martin, C. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nat. Biotechnol. 22, 746–754 (2004).
Bartley, G. E. et al. Transcriptional regulation of chlorogenic acid biosynthesis in carrot root slices exposed to UV-B light. Plant Gene 7, 1–10 (2016).
Wang, H. et al. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc. Natl Acad. Sci. USA 107, 22338–22343 (2010).
Zha, L. et al. DNA methylation influences chlorogenic acid biosynthesis in Lonicera japonica by mediating LjbZIP8 to regulate phenylalanine ammonia-lyase 2 expression. Front. Plant. Sci. 8, 1178 (2017).
Zhang, J. et al. Genome-wide association studies and expression-based quantitative trait loci analyses reveal roles of HCT2 in caffeoylquinic acid biosynthesis and its regulation by defense-responsive transcription factors in Populus. N. Phytol. 220, 502–516 (2018).
Hiruma, K. Roles of plant-derived secondary metabolites during interactions with pathogenic and beneficial microbes under conditions of environmental stress. Microorganisms 7, 362 (2019).
Wink, M. Plant secondary metabolites modulate insect behavior-steps toward addiction? Front. Physiol. 9, 364 (2018).
McLean, S. & Duncan, A. J. Pharmacological perspectives on the detoxification of plant secondary metabolites: implications for ingestive behavior of herbivores. J. Chem. Ecol. 32, 1213–1228 (2006).
Germanà, M. A. Anther culture for haploid and doubled haploid production. Plant Cell Tissue Organ Cult. 104, 283–300 (2011).
Kang, X. Y. Heating treatment device for non-separated branches and buds of trees. CN Patent No. 200610113448.X (2006).
Feuillet, C., Leach, J. E., Rogers, J., Schnable, P. S. & Eversole, K. Crop genome sequencing: lessons and rationales. Trends Plant Sci. 16, 77–88 (2011).
Yahav, T. & Privman, E. A comparative analysis of methods for de novo assembly of hymenopteran genomes using either haploid or diploid samples. Sci. Rep. 9, 6480 (2019).
Simpson, J. T. & Pop, M. The theory and practice of genome sequence assembly. Annu. Rev. Genomics Hum. Genet. 16, 153–172 (2015).
Bourque, G. et al. Ten things you should know about transposable elements. Genome Biol. 19, 199 (2018).
Chuong, E. B., Elde, N. C. & Feschotte, C. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017).
Momohara, A. Floral and paleoenvironmental history from the late Pliocene to middle Pleistocene in and around central Japan. Palaeogeogr. Palaeoclimatol. Palaeoecol. 108, 281–293 (1994).
Cerling, T. E. in C4 Plant Biology (eds Sage, R. F. & Monson, R. K.), 445–469 (Academic Press, 1999).
Grandbastien, M. A. Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 3, 181–187 (1998).
Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61 (2013).
Panchy, N., Lehti-Shiu, M. & Shiu, S. H. Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316 (2016).
Vranova, E., Coman, D. & Gruissem, W. Structure and dynamics of the isoprenoid pathway network. Mol. Plant 5, 318–333 (2012).
Putter, K. M., Van Deenen, N., Unland, K., Prufer, D. & Schulze Gronover, C. Isoprenoid biosynthesis in dandelion latex is enhanced by the overexpression of three key enzymes involved in the mevalonate pathway. BMC Plant Biol. 17, 88 (2017).
Koyama, T. Molecular analysis of prenyl chain elongating enzymes. Biosci. Biotechnol. Biochem. 63, 1671–1676 (1999).
Epping, J. et al. A rubber transferase activator is necessary for natural rubber biosynthesis in dandelion. Nat. Plants 1, 15048 (2015).
Takahashi, S. & Koyama, T. Structure and function of cis-prenyl chain elongating enzymes. Chem. Rec. 6, 194–205 (2006).
Schmidt, T. et al. Characterization of rubber particles and rubber chain elongation in Taraxacum kok-saghyz. BMC Biochem. 11, 11 (2010).
Hillebrand, A. et al. Down-regulation of small rubber particle protein expression affects integrity of rubber particles and rubber content in Taraxacum brevicorniculatum. PLoS ONE 7, e41874 (2012).
Yamashita, S. et al. Identification and reconstitution of the rubber biosynthetic machinery on rubber particles from Hevea brasiliensis. Elife 5, e19022 (2016).
Tangpakdee, J. et al. Structure and biosynthesis of trans-polyisoprene from Eucommia ulmoides. Phytochemistry 45, 75–80 (1997).
Liang, P. H., Ko, T. P. & Wang, A. H. J. Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 269, 3339–3354 (2002).
Liu, J. et al. The Chromosome-based rubber tree genome provides new insights into spurge genome evolution and rubber biosynthesis. Mol. Plant 13, 336–350 (2020).
Pendleton, M. et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Ghurye, J., Pop, M., Koren, S., Bickhart, D. & Chin, C. S. Scaffolding of long read assemblies using long range contact information. BMC Genom. 18, 527 (2017).
Acknowledgements
We thank Drs. Jianbo Xie, Jianfeng Mao and Shihui Niu from Beijing Forestry University for additional help. This work was supported by the Special Funds for Beijing Common Construction Project and Key R & D Plan of Hebei Province (19226339D). We thank Dr. Jenifer Sanders for proofreading the manuscript.
Author information
Authors and Affiliations
Contributions
Y.L. and X.K.conceived and designed the study; Y.L., J.Y., K.D., Y.Z., T.Q., Z.L., Y.R., and L.S. contributed to the sample preparation; J.L. identified and analyzed transcription factors; Y.L, H.W., and X.K. wrote and revised the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Wei, H., Yang, J. et al. High-quality de novo assembly of the Eucommia ulmoides haploid genome provides new insights into evolution and rubber biosynthesis. Hortic Res 7, 183 (2020). https://doi.org/10.1038/s41438-020-00406-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-020-00406-w
This article is cited by
-
Whole-genome sequencing in medicinal plants: current progress and prospect
Science China Life Sciences (2024)
-
Genomic insight into domestication of rubber tree
Nature Communications (2023)
-
Whole genome re-sequencing reveals the genetic diversity and evolutionary patterns of Eucommia ulmoides
Molecular Genetics and Genomics (2022)
