
Abstract
Leaf rust is one of the important diseases limiting global wheat production and productivity. To identify quantitative trait nucleotides (QTNs) or genomic regions associated with seedling and adult plant leaf rust resistance, multilocus genome-wide association studies (ML-GWAS) were performed on a panel of 400 diverse wheat genotypes using 35 K single-nucleotide polymorphism (SNP) genotyping assays and trait data of leaf rust resistance. Association analyses using six multi-locus GWAS models revealed a set of 201 significantly associated QTNs for seedling and 65 QTNs for adult plant resistance (APR), explaining 1.98–31.72% of the phenotypic variation for leaf rust. Among these QTNs, 51 reliable QTNs for seedling and 15 QTNs for APR were consistently detected in at least two GWAS models and were considered reliable QTNs. Three genomic regions were pleiotropic, each controlling two to three pathotype-specific seedling resistances to leaf rust. We also identified candidate genes, such as leucine-rich repeat receptor-like (LRR) protein kinases, P-loop containing nucleoside triphosphate hydrolase and serine-threonine/tyrosine-protein kinases (STPK), which have a role in pathogen recognition and disease resistance linked to the significantly associated genomic regions. The QTNs identified in this study can prove useful in wheat molecular breeding programs aimed at enhancing resistance to leaf rust and developing next-generation leaf rust-resistant varieties.
Similar content being viewed by others


Introduction
Wheat is one of the major staple food crops contributing to 20% of the dietary calories to humans globally (Shiferaw et al. 2013). It is affected by several foliar diseases, of which rust diseases, including leaf or brown rust, stem or black rust and stripe or yellow rust are important. Leaf or brown rust (LR) caused by Puccinia triticina Eriks. (Pt) is considered one of the most devastating diseases of wheat, as it causes a substantial reduction in yield by reducing the number of kernels per ear and kernel weight (Bolton et al. 2008; Marasas et al. 2004). Leaf rust infection during the early stages of crop growth can cause more than 50% yield losses (Huerta-Espino et al. 2011). It is widespread throughout wheat-growing regions of the world, mainly due to its wide adaptability and causes various levels of damage (McCallum et al. 2012). In India, LR is commonly prevalent in all wheat growing regions, as the disease is favored by intermediate temperatures. The development of disease-resistant wheat cultivars is considered the most effective and economic strategy for protecting against yield losses due to LR.
To date, 80 genes for resistance to leaf rust (Lr) have been cataloged (McIntosh et al. 2020). However, not all of them have been successfully utilized in wheat breeding programs. Most of the Lr genes identified thus far confer qualitative resistance, which is race-specific and results in a hypersensitive response or cell death upon infection. This type of resistance is known as vertical or seedling resistance (SR), where the resistance gene in the host and the avirulence gene in the pathogen develop incompatible interactions (Flor 1956). However, such resistance is short-lived due to its breakdown by the new virulent races. Frequent shifts in the virulence pattern of the pathogen have often led to the repeated breakdown of major R genes (McDonald and Linde 2002). In contrast, adult plant resistance genes are race non-specific genes conferring quantitative resistance known as horizontal resistance. This resistance is controlled by genes with small effects providing long-term resistance (Johnson 1984) and reduced susceptibility. The majority of Lr resistance genes reportedly confer leaf rust resistance in the seedling stage and are race-specific (McIntosh et al. 2017), except for a few genes, such as Lr34/Yr18/Sr57, Lr46/Yr29/Sr58, Lr67/Yr46/Sr55 and Lr68 (McIntosh et al. 2017), which are adult plant resistance genes and are race non-specific. The combination of both seedling (SR) and adult plant resistance (APR) genes can strengthen the durability of resistance to LR (Ellis et al. 2014; Kumar et al. 2019). Molecular marker-assisted selection schemes have been applied to combine SR and APR genes to develop improved resistance to LR in wheat.
The constant selection pressure imposed on the pathogen along with long-term deployment of varieties with single major resistance genes has led to the emergence of new races (Kolmer 2013). This has necessitated the need to hunt for new sources of resistance genes to enhance resistance to emerging races (Kertho et al. 2015). Several studies have demonstrated that potential sources for new LR resistance exist in wheat landraces, wild relatives, etc. (Reif et al. 2005). Fortunately, diversity in the form of cultivated and wild accessions, including landraces, are stored as collections in the Genebank. Such diversity needs to be harnessed for the identification of novel resistance that can be utilized in wheat improvement.
Numerous QTLs for LR have been identified thus far using the traditional QTL mapping methodologies that identify the underlying genetic variation using bi-parental mapping populations (Bokore et al. 2017). However, there are several limitations associated with QTL interval mapping, including the long time required for the development of bi-parental mapping populations, less diversity sampled, and low resolution (Zhu et al. 2008; Mir et al. 2012; Gupta et al. 2012). The genome-wide association approach provides a good alternative approach for the identification of novel QTLs/genes for different traits, including leaf rust resistance in wheat (Mir et al. 2012; Kulwal et al. 2022). The availability of genome-wide genotyping data along with powerful statistical models has led to the widespread use of association mapping. In comparison to bi-parental linkage mapping, association mapping is a less time-consuming approach for the discovery of genes/QTL or marker-trait associations (MTAs). In general, association studies are based on germplasm collections representing well-characterized populations that exhibit variation for single or multiple traits of interest (Hall et al. 2010; Zhao et al. 2011; Riedelsheimer et al. 2012).
The identification of QTLs in a natural population using GWAS is possible at higher mapping resolution, as it harnesses all historical recombinations accumulated over generations (Yu and Buckler 2006; Semagn et al. 2010; Brachi et al. 2011; Mir et al. 2012; Kulwal et al. 2022). Population structure is taken into account by including them as fixed-effect covariates in the association model, as it can cause spurious associations in genome-wide association studies (Segura et al. 2012). Thus, GWAS can be considered an effective alternative to bi-parental linkage mapping and can be performed on any existing panel with diversity for the trait. From this perspective, wheat germplasm collections maintained in gene banks could prove valuable genetic resources for hunting genes/genomic regions using an association genetics approach. Several traits in wheat, such as grain yield and yield-contributing traits (Sukumaran et al. 2018; Mir et al. 2021), pre-harvest sprouting resistance (Zhou et al. 2017; Jaiswal et al. 2012), stem rust resistance (Muleta et al. 2017a; Edae et al. 2018; Kumar et al. 2020a), stripe rust resistance (Pradhan et al. 2020; Juliana et al. 2018; Muleta et al. 2017b; Kumar et al. 2020a), eyespot disease resistance (Zanke et al. 2017), agro-morphological traits (Kumar et al. 2020b; Sheoran et al. 2019) etc., have been successfully dissected using GWAS. Few studies on association mapping for LR resistance in bread wheat (Kertho et al. 2015; Aoun et al. 2016; Juliana et al. 2018; Sapkota et al. 2019a; Kumar et al. 2020a; Joukhadar et al. 2017) and durum wheat (Aoun et al. 2019) have been previously reported.
Several statistical models have been used for association mapping involving different approaches (Kaler et al. 2020 and Wang et al. 2016a). Single-locus genome scans performed using ordinary mixed models do not adequately account for large effect loci; hence multi-locus (ML-GWAS) models were proposed as a potential tool for addressing this issue (Rakitsch et al. 2013; Segura et al. 2012). The ML‐GWAS models are considered efficient and more reliable than single locus (SL‐GWAS) models for mapping genomic regions because in ML‐GWAS models, all‐marker effects are simultaneously estimated. Moreover, unlike SL‐GWAS models, these do not require testing of identified associations using stringent multiple testing corrections that generally result in rejection of significant associations (Zhang et al. 2019). They also have higher power to detect significant marker-trait associations than single-locus models (Wang et al. 2016a). The multi-locus mixed model (MLMM) introduced by Segura et al. (2012) uses stepwise regression to incorporate the most influential markers as cofactors and has been used successfully in several association mapping studies to date (Danakumar et al. 2021; Lipka et al. 2013; Sauvage et al. 2014; Vaughn et al. 2014). Some commonly used multi-locus models include multi-locus random-SNP-effect mixed linear model (mrMLM (Wang et al. 2016a)), iterative modified-sure independence screening Expectation-Maximization-Bayesian least absolute shrinkage and selection operator (ISIS EM-BLASSO (Tamba et al. 2017)), fast multi-locus random-SNP-effect efficient mixed model analysis (FASTmrEMMA (Wen et al. 2018)), polygenic-background-control based least angle regression plus empirical Bayes (pLARmEB (Zhang et al. 2017a)) and fast multi-locus random-SNP-effect mixed linear model (FASTmrMLM (Tamba and Zhang 2018)). In the present study, we used six ML-GWAS models on the association mapping panel to identify novel genomic regions associated with both seedling and adult plant stages of leaf rust resistance. To the best of our knowledge, this is the first attempt to identify significantly associated genomic regions/candidate genes to leaf rust pathotypes in wheat using multiple ML-GWAS models.
We performed a GWAS to identify genomic regions responsible for resistance to LR from a diverse set of germplasms comprising advance breeding lines (189), landraces (186), and cultivars (25) using the 35 K single nucleotide polymorphism (SNP) array-based on seedling and adult plant stage leaf rust responses under controlled and natural field conditions, respectively. The identification of wheat genomic regions that influence LR resistance will improve our understanding and facilitate the development of LR resistant wheat cultivars.
Materials and methods
Germplasm/Plant materials utilized
A total of four hundred (400) bread wheat accessions comprising advance breeding lines (189), landraces (186) and cultivars (25) obtained from ICAR-National Bureau of Plant Genetic Resources (NBPGR) were used in the association mapping panel for this study. This rust association mapping panel was evaluated against Puccinia triticina for leaf rust both at the seedling stage in controlled conditions and at the adult plant stage in the field.
Seedling resistance evaluation
Wheat accessions were inoculated with four predominant pathotypes of leaf rust, viz., 12-5 (29R45 = FHTPM), 77-5 (121R63-1=THTTM), 77-9 (121R60-1 = MHTKL) and 104-2 (21R55 = PHTTL) under glasshouse conditions at the ICAR-Indian Institute of Wheat and Barley Research (IIWBR), Regional Station, Shimla, India. These four pathotypes were selected based on the prominence and virulence. The seedling response test (SRT) was performed twice (Y1 and Y2) as mentioned by Bhardwaj (2011). Among the pathotypes present, 77-5 and 77-9 are more virulent and prevalent than pathotypes 12-5 and 104-2 in the field, so the same pathotypes were used in the seedling test. The disease response was recorded as infection type (IT), 14 days post-inoculation using a 0–4 scale (Stakman et al. 1962). The scores were further linearized to a 0–9 scale (Ziems et al. 2014; Riaz et al. 2016) as follows: 0; = 0;- = 0.5 = 1, 1- = 1.5, 1 = 2, 1 += 3, 2- = 4, 2 = 5, 2 + = 6, 3- = 6.5, 3 = 7, 3 + = 8 and 4 = 9. Genotypes with an IT score of 0 to <3, >3 to <6, and >6 to 9 were considered resistant, moderate and susceptible, respectively.
Adult plant evaluations
Adult plant evaluation for LR was carried out at the ICAR-Indian Agricultural Research Institute (IARI), Regional Station, Wellington (11°22′47.5″N, 76°46′26.1″E), Tamil Nadu, India, which is the hot spot for leaf rust, stem rust, and powdery mildew diseases of wheat. Wellington located in the southern Indian Nilgiri hills is gifted with a natural environment for harboring pathotypes of wheat rust throughout the year. Phenotypic evaluation under natural conditions for LR was performed for two consecutive years (2018–19 and 2019–20) in four seasons (summer - 2018 & 2019; winter - 2018–19 & 2019–20) and each environment was represented as S_E1, W_E1, S_E2 and W_E2 based on the season and location.
In each season, accessions were planted in a non-replicated augmented block design including three resistant (DPW621-50, HW2022 and HW5075) and susceptible checks (Morocco, Lal Bahadur and WL711). Each accession was planted in a single 1 m row, with a spacing of 20 cm between the rows. Resistant and susceptible checks were planted after every 20 rows of test accessions. Moreover, susceptible checks were planted around each plot for a continuous supply of leaf rust inoculum.
LR infections were scored thrice from the early to advance phases of disease development in 7–10 day intervals coinciding with the beginning of anthesis (Zadoks 60) to the dough development stage (Zadoks 85). Recordings of LR reactions against Puccinia triticina were based on two measures: disease severity (DS) and infection response (IR). DS was measured using the modified Cobb scale (Peterson et al. 1948) as an estimation of percentage coverage (0–100) of LR pustules (uredinia) on the leaves. IR based on host response to leaf rust (pustule size) was measured according to Loegering (1959), 0—No visible infection; R (Resistant)—Visible chlorosis or necrosis, no uredia present; MR (Moderately resistant) - Small uredia are present and surrounded by either chlorotic or necrotic areas; MX (Intermediate) - variable sized uredia, some with necrosis or chlorosis; MS (Moderately susceptible): Medium-sized uredia are present and possibly surrounded by chlorotic area, S (Susceptible): Large uredia are present, generally with little or no chlorosis or necrosis. IR data were converted to a numeric scale of 0 to 1 (Roelfs et al. 1992), where Immune = 0.0, Resistant (R) = 0.2, Moderately Resistant (MR) = 0.4, Mixed response (MX) = 0.6, Moderately Susceptible (MS) = 0.8, and Susceptible (S) = 1.
For association mapping analysis, disease severity (DS) and infection type (IR) were combined into a single value known as coefficient of infection (COI), which is the product of DS and IR (Loegering 1959; Roelfs et al. 1992). The COI values ranged from 0–100, where COI values of 0–20 were considered resistant, 20–60 as moderate, and >60 as susceptible. COI values were used for detecting QTNs (Auon et al. 2016; Kumar et al. 2020a).
Statistical analysis of phenotypic data
The statistical analysis of the phenotypic data was analyzed by statistical analysis system (SAS) software v9.4 (SAS Institute Inc. 2017). The Shapiro-Wilk test was conducted (PROCUNIVARIATE) to determine if the phenotypic data for each pathotype were normally distributed. Furthermore, depending on the result from the normality test, Levene’s or Bartlett’s test (Levene 1961; Snedecor and Cochran 1989) was performed to check the homogeneity of data among the experiments. Broad-sense heritability of the pathotypes was estimated using the restricted maximum likelihood (REML) method. Pearson’s correlation in the phenotypic data of each pathotype was calculated using the program R. Descriptive statistics and frequency distribution were also presented to understand the variability of pathotypes in the association panel.
DNA extraction and SNP genotyping
The genomic DNA from all 400 lines was extracted separately from 15-day old seedlings by following the CTAB procedure (Doyle 1990). Genotyping of the association panel was performed using a 35 K Axiom Wheat Breeders Array according to the method described by Affymetrix (Axiom 2. 0 Assay for 384 samples P/N 703154 Rev. 2) for wheat resulting in 35,143 SNPs. All SNP markers with minimum allele frequency (MAF < 0.1), maximum missing site per SNP > 20%, and call rate >90% were excluded from further downstream analysis. Finally, a total of 18,932 polymorphic SNPs were used in GWAS analysis.
Population structure and linkage disequilibrium
To examine the population structure (Q) present in the wheat panel used in the current study, STRUCTURE v2.3.4 (Pritchard et al. 2000) was used. K value (i.e., No. of sub-groups/sub-populations) was assumed to range from 1–10 with a burn-in of 10,0000 iterations, and 20,0000 Monte Carlo Markov chain (MCMC) replicates were provided as initial estimates of parameters to the STRUCTURE tool. Furthermore, a web-based utility Structure Harvester (http://taylor0.biology.ucla.edu/structureHarvester/), which is based on the approach of Evanno et al. (2005), was used to estimate the optimum number of sub-groups (K) in the panel. This estimated number of sub-groups in the genotype panel is based on maximum likelihood estimates of the proportion of each sample derived from each of the K sub populations. Population structure Q was also obtained for further investigation and used in the GWAS analysis.
Intra-chromosomal LD between all possible pair-wise comparisons of SNPs was estimated as squared allele frequency correlation (r2) using TASSELv5.0 (Bradbury et al. 2007). The background LD in the wheat AM panel was estimated to identify the critical distance for LD decay. The average pattern of genome-wide LD decay over physical distance was determined by constructing a scatter plot of r2 values against the corresponding physical distance among the markers. Furthermore, the extent of LD decay was estimated using the Locally Weighted Scatter-plot Smoother (LOESS) model (Cleveland 1979). The critical r2 value that shows the area beyond which LD is due to true physical linkage was determined using the 95th percentile of the square root of transformed r2 data of unlinked markers (Breseghello and Sorrells 2005). Furthermore, the intersection of the LD decay curve was observed at r2 = 0.157 and used as a threshold.
Genome-wide association analyses
Association analyses were performed using six ML-GWAS models, including mrMLM, FASTmrMLM, FASTmrEMMA, pLARmEB, ISIS EM-BLASSO, and pkWmEB. The FASTmrMLM method is relatively faster with higher statistical power and accuracy in estimating QTNs compared to mrMLM (Tamba and Zhang 2018). ISIS EM-BLASSO can detect significant associations with the highest robustness and accuracy compared to mrMLM and FASTmrEMMA (Tamba et al. 2017). The pLARmEB ML-GWAS model integrates least angle regression with empirical Bayes, which enables a more accurate estimation of significantly associated QTNs in polygenic backgrounds (Zhang et al. 2017a). The above six models were implemented in the R package mrMLM v4.0 (https://cran.r-project.org/package=mrMLM) with default parameters. The QTNs with an LOD score of ≥3.00 were considered significantly associated with both seedling and adult plant resistance to leaf rust. Further, SNP markers that were identified in at least two models, were designated as reliable leaf rust-associated QTNs. Similarly, QTNs that were repeatedly detected in three or more than three models and showed phenotypic variation (R2 > 10%) designated as major QTNs.
Identification of potential candidate genes
To identify the potential candidate genes, SNPs (probe sequences) that were significantly associated with seedling and adult plant stage resistance to leaf rust pathotypes were searched using the Basic and Local Alignment Search Tool against the reference genome of Triticum aestivum using ensemble Plants (http://plants.ensembl.org/index.html) with default parameters. Expressed transcripts around significantly associated QTNs were identified, and their nucleotide sequences were extracted. Annotation of expressed transcripts was performed using BLAST2GO software (Conesa et al. 2005).
Results
Statistical data analysis
The frequency distribution of the infection types (ITs) produced by the four different leaf rust pathotypes, i.e., 12-5, 77-5, 77-9 and 104-2, in the association mapping panel is shown in Fig. 1. The Shapiro–Wilk test for normality showed that the phenotypic data of all four studied pathotypes of leaf rust significantly deviated from a normal distribution. Therefore, Levene’s test was performed to test for homogeneity within experiments. The Levene’s test results indicated that the phenotypic variance of the data within experiments was homogenous (P = 0.63–0.91) for all four studied pathotypes (Supplementary Table S1). Therefore, the overall mean value was calculated for each wheat genotype and used in the GWAS. Moreover, it was also observed that the Pearson’s correlation coefficients (r) among all studied pathotypes for both seedling and adult plant stages were highly significant (p < 0.0001) (Fig. 2a, b).

The x-axis is the linearized disease scale, and the y-axis is the number of wheat genotypes of the four pathotypes.

a At seedling stageand b at adult plant stage.
For adult plant resistance, the distribution of phenotypic values was visualized using violin plots (Supplementary Fig. S2). The broad-sense heritability of the leaf rust infection types ranged from 0.98 to 0.99 for the four different pathotypes at the seedling stage and 0.99 at the adult plant stage, indicating that most of the phenotypic variation was explained by the genotypes.
Phenotypic evaluation for leaf rust
The avirulence/virulence profile of the pathotypes for leaf rust is presented in Supplementary Table (S2). Seedling reactions showed significant differences among accessions for each pathotype. However, no significant differences in IT scores were observed in resistant, moderate, and susceptible accessions between years. The majority of the wheat accessions in the AM panel displayed susceptible IT scores for all pathotypes except 12-5 (29R45). For pathotypes 12-5 (29R45), 77-5 (121R63-1), 77-9 (121R60-1) and 104-2 (21R55), resistant ITs were observed in 42%, 9%, 13% and 9% of the accessions, respectively (Supplementary Table S3). Pathotype 77-5 (121R63-1) was the most virulent, followed by 77-9 (121R60-1) and 104-2 (21R55) pathotypes on 84%, 74%, and 71% of susceptible accessions, respectively, while pathotype 12-5 (29R45) was avirulent on 35% of susceptible accessions. Only 27 accessions (6.75%) showed resistance to all pathotypes, consisting of advanced breeding lines (17), landraces (6), and cultivars (4) (Supplementary Table S4). Among the pathotypes, correlation coefficients ranged from low to medium.
The adult plant response of the AM panel under field conditions for four seasons (Summer_E1, Winter_E1, Summer_E2 and Winter_E2) was evaluated in terms of disease severity (DS), infection response (IR), and coefficient of infection (COI). However, coefficient of infection (COI) values, which are the product of DS and IR, were used for GWAS analyses. In contrast to SRT, approximately 75% of the accessions displayed resistance reactions in all four environments. The COIs of 77%, 74%, 76% and 73% of the accessions showed resistance reactions to leaf rust in the S_E1, W_E1, S_E2, and W_E2 environments, respectively. Moderate and susceptible reactions were observed in 22 to 25% and 0.5 to 1% accessions, respectively. The presence of APR genes in the background of the accessions provided resistance in the adult plant stage compared to the seedling stage (Supplementary Table S5). Twenty-six accessions (6.5%) showed resistance reactions to leaf rust at both the seedling and adult plant stages, comprising advanced breeding lines (17), landraces (5), and cultivars (4) (Supplementary Table S4 and Table S9). A high correlation (0.9) was observed among environments for the leaf rust response (S_E1, W_E1, S_E2 and W_E2).
Population structure and LD decay
Four hundred (400) wheat accessions were genotyped for the AM panel using a 35 K Axiom SNP array. After ensuring quality checks, such as the removal of monomorphic and low-quality SNPs, a total of 18,932 SNPs with their physical positions were used for GWAS analyses. The physical positions of these SNPs were obtained through BLAST search using wheat genome version IWGSC v2.0. Out of 18,932 mapped SNPs, 6036 were mapped to the A sub-genome, 7191 to the B sub-genome, and 5705 to the D sub-genome. The number of SNPs mapped on individual chromosomes ranged from 377 (Chr4D) to 1307 (Chr2B) (Table 1). The chromosomal level distribution of SNPs on three sub-genomes showed (Supplementary Fig. S1) that the A sub-genome had the highest number of SNPs on Chr2A (1072), followed by Chr1A (1042) and Chr7A (984); the B sub-genome had the maximum SNPs on Chr2B (1307), followed by Chr5B (1248) and Chr1B (1155), whereas the D sub-genome had the maximum number of SNPs on Chr2D (1168), followed by Chr1D (1028) and Chr5D (892). However, to avoid homologous SNPs (which might be present across the three wheat sub-genomes), only 18,932 SNPs with exact physical locations were considered for further analysis.
Population structure analysis categorized the 400 accessions into 4 different sub-populations, SP1, SP2, SP3 and SP4, based on the ∆K method, consisting of 57, 16, 130, and 197 accessions, respectively. Sub-population 4 (SP4) was the largest among the four sub-populations with 49.2% of the total accessions from the studied panel, followed by SP3, SP1, and SP2. Individuals of each population could be further categorized into two groups, i.e., pure and admixture types. Populations comprising ≥ 0.8 of the member proportions were considered pure and others were considered admixtures. Considering this criterion, the compositions of the four sub-populations were as follows (Supplementary Fig. S3 A): SP1: 36% pure and 64% admixtures, SP2: 87% pure and 13% admixtures, SP3: 48% pure and 52% admixtures, and SP4: 11% pure and 89% admixtures. Out of the four sub-populations, SP4 consisted of a higher proportion of resistant accessions than the others. Principal component analysis (PCA) was also performed, where PC1 explained 32.8% of the genetic variance, while PC2 explained 13.0%. The PC analysis scatter plot (Supplementary Fig. S3 B) showed that the first and second PCs distinguished the four sub-populations of genotypes originating from different regions, which also supported the results of the population structure analysis.
A total of 18,932 markers selected for association analysis were used to measure pairwise LD between markers. LD was measured as the squared allele frequency correlation (R2) between pairs of intra-chromosomal SNP’s with known chromosomal positions. The LD (R2) across 21 wheat chromosomes ranged from 0.088 (Chr4D) to 0.3-1 (Chr1D). The sub-genome LD level estimate for the A sub-genome was 0.157, the B sub-genome was 0.154 and the D sub-genome was 0.147. A detailed summary of markers, including chromosome distribution, average LD score, and other associated statistics is presented in Table 1. The background LD of 0.157 was considered the threshold cut-off for estimating LD decay. It was observed that in the selected wheat AM panel, LD decayed the fastest in the A sub-genome followed by B and D. In the A sub-genome, the R2 value for the marker pairs was 0.157 (used as the threshold) at 1.9 Mb compared to 2.8 Mb in D and 4.1 Mb in the B sub-genome (Supplementary Fig. S4).
Genome-wide association analysis
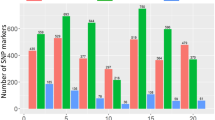
Identification of defence genes and genomic regions associated with four different pathotypes at the seedling stage and coefficient of infection at the adult plant resistance stage were performed using various models. In using six different ML-GWAS models, a total of 201 QTNs were identified for resistance to 4 different leaf rust pathotypes at the seedling stage (Supplementary Table S6 and Fig. 3 a–c), and 65 QTNs for APR were distributed on 21 chromosomes (Supplementary Table S6). The phenotypic variation (PVE) estimated (R2) for both seedling and APR ranged from 1.98% to 31.72%, indicating that leaf rust pathotypes are controlled by multiple loci with small to moderate effects. This also shows the complex genetic control of these pathotypes at an early stage of crop growth. Among the six ML-GWAS models used in our analysis, the pLARmEB model was the most powerful, which revealed the maximum number of associations QTNs (55), whereas FASTmrEMMA was the least powerful, as it detected the lowest number of QTNs (24). For the seedling stage, identified QTNs were distributed on 19 wheat chromosomes, indicating good coverage of the A, B, and D sub-genomes. The highest number of QTNs was identified on chromosome 3D (35), followed by 5D (22), 3A (21), 3B (18), 1D (11), and 6D (11). Out of 201 QTNs, 51 were resistant to 4 different pathotypes (i.e., 12-5, 77-5, 77-9, and 104-2) that were repeatedly detected by at least two models are summarized in Table 2 and Fig. 4. Furthermore, the pathotype-wise distributions of these 51 reliable QTNs were 12-5 (13), 77-5 (14), 77-9 (13), and 104-2 (11). Furthermore, we compared the locations of identified QTNs in our study with those of previous studies (Supplementary Table S10).

a Number of significant QTNs detected for both seedling and adult plant resistance across six multi-locus GWAS methods. b Number of significant QTNs detected using each of six multi-locus GWAS methods. c Number of QTNs per chromosome.

Different colors indicate different pathotypes, i.e., blue:77-5, green:77-9, red:104-2, purple:12-5 and pink: adult plant resistant (APR).
QTNs for seedling
For pathotypes 12-5, a total of thirteen reliable QTNs were identified in the association panel. (Table 2 and Fig. 5). Out of these thirteen QTNs, QLr.iari-1AL_12-5, QLr.iari-1DS_12-5, QLr.iari-3DL.2_12-5 and QLr.iari-5DL.2_12-5 were detected by at least two models and explained 2.01% to 9.06% of the total phenotypic variation. QLr.iari-3DL.1_12-5 represents the most significant QTN for pathotypes 12-5, with the highest LOD score (9.45).

a For pathotype 12-5, b for pathotype 77-5, c for pathotype 77-9, d for pathotype 104-2, and e for adult plant stage.
Similarly, fourteen reliable QTNs were significantly associated with resistance to the 77-5 pathotype and were distributed on 10 chromosomes, including 1A, 1D, 3A, 3B, 3D, 5D, 6D, 7A, and 7D. The highest number of QTNs was located on the D sub-genome. Out of fourteen, six QTNs, QLr.iari-3AL.1_77-5, QLr.iari-3BL.2_77-5, QLr.iari-3DS.2_77-5, QLr.iari-5DL_77-5, QLr.iari-6DL_77-5, and QLr.iari-7AS_77-5, were identified consistently with the results of three GWAS models and explained phenotypic variation in the range of 0.94–20.76%. Among these six QTNs, QLr.iari-3BL.2_77-5 was found to be common with the results of four GWAS models (i.e., FastMrEMMA, pLARmEB, ISIS EM-BLASSO and pkWmEB), and another QTN QLr.iari-5DL_77-5 was detected through five different GWAS models (i.e., MrMLM, FastMrMLM, FastMrEmma, pLARmEB and ISISEM-BLASSO), which were major QTNs, and the other four were minor. Similarly, the QTN on chromosome 3B (QLr.iari-3BL.2_77-5) was a major QTL (R2 > 10% for at least one method) and also explained the highest phenotypic variance with an LOD value of 3.07–24.27.
For resistance to pathotype 77-9, thirteen reliable QTNs were identified. Of these, 7 QTNs, viz., QLr.iari-1DS_77-9, QLr.iari-2AL.1_77-9, QLr.iari-2AL.2_77-9, QLr.iari-3AL_77-9, QLr.iari-3BL_77-9, QLr.iari-5DL_77-9 and QLr.iari-6DL_77-9, were identified using three different models and distributed on chromosomes 1D, 2A, 3A, 3B, 5D and 6D, respectively. All seven reliable QTNs were major and explained 1.75–31.72% of the phenotypic variation for 77-9. Out of seven, QLr.iari-2AL.2_77-9 was detected through five different models (i.e., MrMLM, FastMrMLM, FastMrEMMA, pLARmEB, and ISIS-EM-BLASSO) and explained 1.99-5.91% of the total phenotypic variance.
Eleven QTNs were significantly associated with resistance to the 104-2 pathotype and were on different chromosomal regions, i.e., 1A, 3A, 3B, 3D, 5A, 5D, 7A and 7B and, which explained phenotypic variation in the range of 0.58–18.45%. Furthermore, QLr.iari-3DL.1_104-2 (FastMrMLM, FastMrEMMA, pLARmEB, ISIS EM-BLASSO and pkWmEB) and QLr.iari-5DL_104-2 (MrMLM, FastMrMLM, FastMrEMMA, pLARmEB, ISIS EM-BLASSO and pkWmEB) QTNs were major and detected by five models and represent a highly reliable locus for resistance to the 104-2 pathotype (Supplementary Fig. S6). Among these two QTNs, QLr.iari-3DL.1_104-2 had profound control of seedling stage resistance to the 104-2 pathotype (R2 = 5.80–18.45%, LOD: 4.13–13.90).
QTNs for adult plant resistance
Out of 65 QTNs identified for APR, 15 QTNs were consistently detected by at least two GWAS models, and the results are presented in Supplementary Table S7. These identified QTNs were distributed over eleven different chromosomal regions (i.e., 1A, 1B, 1D, 2A, 2B, 3A, 3B, 3D, 4A, 4D and 6D) and explained 1.30% to 17.83% of the phenotypic variation. Of these, seven QTNs, QLr.iari-1DL_APR, QLr.iari-2AL_APR, QLr.iari-3AL.2_APR, QLr.iari-3AL.4_APR, QLr.iari-3DL_APR, QLr.iari-4AL.1_APR and QLr.iari-6DL_APR, were found to be highly consistent in four or more GWAS models. Of the seven QTNs, four were major and found to be consistent using five different models (i.e., MrMLM, FastMrMLM, FastMrEMMA, pLARmEB, ISIS EM-BLASSO) and explained a range of phenotypic variance (1.32–20.76%) with LOD values (3.07–7.96). Most QTNs identified were found on the long arm of chromosome 3A and explained 2.64% to 7.60% of the phenotypic variance.
Pleiotropic QTNs associated with multiple pathotypes
Responses to different leaf rust pathotypes are generally correlated, and complex biological mechanisms are involved in the coordination of their expression. The pleiotropic action of genetic loci on different pathotypes or their fixed linkage results in a correlation between pathotypes. A total of three QTNs were found to pleiotropically affect the response to different pathotypes (Table 2). A locus on the long arm of 3D (AX-95226287 and AX-95211729) was associated with two different pathotypes (12-5 and 77-9), and another locus on the long arm of 3A (AX-94480950) was associated with 77-5 and 104-2. We also identified one locus on the long arm of chromosome 3A (genomic region < 10 Mb in size) that contained QTNs for both seedling and adult plant resistance (Table 2 and Supplementary Table S7). For example, the genomic region (739.38–739.55 Mb) on 3AL was associated with two QTNs, QLr.iari-3AL_77-9 for seedling resistance and QLr.iari-3AL.4_APR for adult plant resistance. On chromosome 1AL, two QTNs (QLr.iari-1AL_12-5 and QLr.iari-1AL_APR) for both seedling and adult plant resistance were located within 560.96 and 568.00 Mb regions, respectively. Another genomic region (606.94–608.92 Mb) on 3DL (3 QTNs) was associated with pathotypes 12-5 and 77-5. Similarly, two QTNs, one each for 12-5 (QLr.iari-5DL.1_12-5) and 104-2 (QLr.iari-5DL_104-2), were found within a small genomic region (545.52–550.19 Mb) on 5DL. These identified loci influencing several pathotypes could be potential markers for future marker-assisted breeding (MAS) programs after validation. For a better understanding of the results, chromosome-wise distributions of significant QTNs have also been presented as Manhattan plots (Fig. 5).
Identification of potential candidate genes
Putative candidate genes associated with seedling resistance to pathotypes 12-5, 77-9 and 104-2 belonged to the serine-threonine/tyrosine-protein kinase family, which has a robust role in pathogen recognition and disease resistance (Bhatia et al. 2021; Pradhan et al. 2020; Afzal et al. 2008) (Table 3 and Supplementary Table S8). Similarly, putative candidate genes linked to the markers associated with the response to pathotype 77-5 belonged to the leucine-rich repeat domain superfamily that contributes to pathogen effector recognition (Kumar et al. 2021; Marone et al. 2013; Feuillet et al. 1997). Similarly, 19 drought-induced protein types, zinc-binding domain proteins that are involved in plant disease resistance (Gupta et al. 2012), and regulators of chromosome condensation 1/beta-lactamase-inhibitor protein II, were also functionally annotated for markers associated with the response to pathotype 12-5 (Narayanan et al. 2009). Modulation of defence responses against pathogens was also involved in replication machinery involving DEAD-box ATP-dependent RNA helicase 58, chloroplastic (Li et al. 2008), and a versatile F-box-like domain superfamily protein, master regulator for protein degradation and broadly targeted by miRNAs (Li et al. 2020). Similarly, markers associated with pathotype 77-5 and 77-9 trigger a response against the host by hydrolysis using P-loop-containing nucleoside triphosphate hydrolase (Juliana et al. 2018; Wu et al. 2019) and the tetratricopeptide-like helical domain superfamily. A possible role against seedling resistance was also provided by Isopenicillin N synthase-like, oxoglutarate/iron-dependent dioxygenase and domain of unknown function DUF1618 (Pradhan et al. 2020; Naoumkina et al. 2010; Riaz et al. 2018). Notably, a few more genes, including an amino acid transporter, transmembrane domain, C2 domain superfamily, flavin-containing monooxygenase, aspartokinase, and ubiquitin-specific protease domain, were also identified as potential candidates against diverse sets of pathotypes against leaf rust (Sonawala et al. 2018; Zhang et al. 2013; Serba et al. 2015; Cloutier et al. 2008; Nazeem et al. 2011; Zanke et al; 2017; Pariyar et al. 2016).
Gene annotation for adult plant resistance against leaf rust pathogens covers the most similar plethora of proteins involved in seedling resistance because both shared homology with the signalizing cascade and elicitor response. However, a few additional annotated proteins, including histone acetyltransferase Rtt109/CBP (Saripalli et al. 2020; Sharma et al. 2019), S-formylglutathione hydrolase (Saintenac et al. 2013), NADH-ubiquinone oxidoreductase, 20 Kd subunit (Jain et al. 2021; Song et al. 2011) and a few additional proteins involving AT-hook motif nuclear-localized proteins, alpha/beta hydrolase fold, superoxide dismutase (Cu/Zn)/superoxide dismutase copper chaperone, and subtilisin-like protease SBT1.3 (Jain et al. 2021; Saripalli et al. 2020; Singh et al. 2017; Juliana et al. 2018), have already been screened against leaf rust pathotypes.
Discussion
Wheat germplasm collections are a repository for novel resources of genes, alleles and QTNs against various pathogens because they were collected from widely distributed geographical environments/locations. Although a diverse set of leaf rust resistance genes have been mapped and utilized, pathogens evolve corresponding avr factors against the resistance genes, resulting in the breakdown of resistance. Such a situation demands the identification, diversification, and utilization of new resistance genes to provide durable resistance. Among the gene pools available for the exploitation of genetic resistance, the primary gene pool comprising landraces, breeding lines, cultivars, etc., constitutes a vital source for novel resistance genes, as they can be immediately used in the development of cultivars (Mujeeb-Kazi et al. 2013). Moreover, advances in high-throughput genotyping, phenotyping, next-generation sequencing, bioinformatics tools, etc., have paved the way to access valuable information from genomic databases and to study a large number of germplasms, enabling effective harnessing of the genetic diversity of the germplasm. The association mapping panel of 400 diverse genotypes, comprising advance breeding lines, landraces, and cultivars characterized in the study, showed significant variations in leaf rust resistance on both seedlings in the greenhouse and adult plants in the field. As the panel was subjected to predominant and virulent leaf rust pathotypes at seedling and adult plant stages, we were able to identify significant resistance QTNs, which can be utilized to broaden the genetic base of leaf rust resistance.
Phenotypic variation for leaf rust resistance
Screening of the association mapping panel for leaf rust resistance was performed at the seedling stage under controlled conditions and at the adult plant stage under field conditions. The panel displayed wide variation in the leaf rust response in both seedling and adult plant stages. Phenotypic data of leaf rust reactions did not follow a normal distribution, but were skewed towards susceptibility in seedlings and towards resistance in adult plant stages. The seedling resistance test of the AM panel showed that more than 70% of the accessions were susceptible to three pathotypes (77-5(121R63-1=THTTM), 77-9 (121R60-1 = MHTKL) and 104-2 (21R55 = PHTTL)), while more than 70% of the accessions exhibited resistance reactions in the adult plant stage. The same susceptible checks used in the field and seedling tests exhibited susceptible reactions across environments and years, which indicates that variation in leaf rust reactions among the accessions in the seedling and adult stages is genetic.
The majority of the resistant accessions (except a few) in the field displayed susceptible reactions in the seedling stage, including advance breeding lines (140), landraces (127), and cultivars (16). A higher number of resistant accessions in the adult plant stage across environments coupled with susceptible reactions in the seedling stage indicated the presence of APR QTLs in the panel. Moreover, 26 accessions were resistant at both the seedling and adult plant stages, which may be due to the presence of the major R gene in the background. In recent years, the emphasis on APR genes for rust resistance in wheat improvement programs has been attributed to the presence of APR genes in advance breeding lines and cultivars, but the presence of APR in landraces is interesting. APR QTLs among landraces may be known or novel, which can diversify leaf rust resistance. Previous studies by Aoun et al. (2016); Sapkota et al. (2019b); Kumar et al. (2020a) and Joukhadar et al. (2020) utilized landraces for harnessing novel leaf rust resistance alleles.
Population structure and linkage disequilibrium
We considered 18,932 SNPs on different chromosomes and observed that sub-genome B had the highest marker density, followed by sub-genomes A and D. A similar pattern in which sub-genome B had the highest marker density among the sub-genomes was also observed previously (Kumar et al. 2020a). The rate of LD decay was faster in genome A than in genomes B and D, which was in concurrence with other studies (Chao et al. 2010; Voss‐Fels et al. 2015; Pradhan et al. 2020).
For any breeding program, genetic diversity is the key factor, therefore, determination of the extent of genetic diversity and population structure are the foremost requirements for utilizing plant genetic resources in breeding programs and genetic studies (Atwell et al. 2010). The AM panel comprised genotypes from diverse geographic regions, of 400 genotypes: 258 were from the northern part of India (Punjab, Haryana, Uttar Pradesh, Uttarakhand, and Himachal Pradesh), 67 were from other parts of India, such as the Central zone (Rajasthan, Madhya Pradesh and Gujarat) and the southern part of India (Karnataka, Tamilnadu and Andhra Pradesh), 59 were from CIMMYT, Mexico, and 16 other genotypes were exotic. Among the indigenous germplasms, 50% of the accessions were landraces or indigenously collected germplasm lines. Different analysis methods (model-based population structure, principal component and cluster analyses) agreed with a common consensus and indicated the presence of four sub-groups (K = 4) that also broadly agree with the geographic origins of the genotypes (Kumar et al. 2020a). Significant admixture was noticed, which might be due to the sharing of the germplasm across breeding programs. We observed that 70% of SP1 included germplasm of Indian origin and were either landraces or breeding lines or released cultivars. Out of 130 genotypes from SP3, 76 accessions were indigenous landraces, while the rest of the accessions were cultivars (23) and breeding lines (31). SP2 contained only 16 accessions, of which eight each were landraces and breeding lines. SP4 had 80% landraces/indigenously collected germplasm of Indian origin and 20% cultivar and breeding lines.
Comparison of identified QTNs with previously published Lr genes/QTNs
Genome-wide association studies are a powerful approach in plants to detect QTNs associated with multiple pathotypes. Genome-wide association studies have been successfully used in association panels to detect several genomic loci/regions conferring resistance to leaf rust at seedling and adult plant growth stages (Kertho et al. 2015; Aoun et al. 2016; Turner et al. 2017; Riaz et al. 2018). Our study detected a total of 51 reliable QTNs for seedling resistance to leaf rust. These QTNs were distributed mostly all over the wheat chromosomes except for a few chromosomes, such as Chr4A, Chr4B, Chr4D, Chr5B, and Chr6A, for the four different pathotypes. These QTNs further need to be validated before use in any future breeding programs. Previous studies have also identified Lr genes/QTNs distributed on almost all wheat chromosomes, as both major and minor genes are known to be responsible for conferring leaf rust resistance at both seedling and adult plant stages (Sapkota et al. 2019b). Furthermore, we compared the locations of identified QTNs in this study with those of previous studies. For some of the QTNs, comparisons across different studies were difficult due to the differences in the marker platforms, mapping populations, and the lack of a consensus map to compare positions. For such QTNs, only chromosome arm positions (short/long) were used for comparison in our analysis. Novel associations for leaf rust resistance identified in the current study along with previously reported ones are discussed below.
Of the 51 QTNs detected at the seedling stage through ML-GWAS, QLr.iari-1AL_12-5 and QLr.iari-1AL_104-2 were found to be significant for resistance to the 12-5 and 104-2 pathotypes, respectively, which were located on the long arm of 1A at 560.95 to 577.47 Mb chromosomal region, which is near the IWA1557 marker (Turner et al. 2017) and QLr.ags-1AL QTL (Sapkota et al. 2019a). Two QTNs, QLr.iari-1DS_12-5 and QLr.iari-1DS_77-9, associated with the 12-5 and 77-9 pathotypes located on chromosome 1DS are near the TSD276-2 gene (Dinkar et al. 2020). The other two QTNs, QLr.iari-2AL.1_77-9 and QLr.iari-2AL.2_77-9, were associated with the 77-9 pathotype and located at 709.62 and 728.40 Mb, respectively, on the long arm of chromosome 2A were present near the IWA5092 marker (Turner et al. 2017).
QTN, QLr.iari-2BL_12-5 detected on chromosome 2BL and associated with the 12-5 pathotype are in the vicinity of the IWA2025, IWA207 and IWA2509 markers (Turner et al. 2017). Similarly, QLr.iari-2BS_77-9 QTN was found at 725.77 Mb on the short arm of chromosome 2B, which is near the IWA4894 marker (Turner et al. 2017). Seven known Lr genes and two QTLs, viz., Lr13 (Dyck et al. 1966), Lr16 (McCartney et al. 2005), Lr23 (McIntosh and Dyck 1975), Lr48 (Bansal et al. 2008), Lr73 (Park et al. 2014), LrZH22 (Wang et al. 2016b), LrA2K (Sapkota et al. 2019a), Q.Lr.cimmyt-2BS (Rosewarne et al. 2012), and QLr.hebau-2BS (Zhang et al. 2017b), were also identified and located on the short arm of chromosome 2B in the vicinity of QTN, QLr.iari-2BS_77-9. Of these seven known genes, Lr13 and Lr48 are APR genes reported in the wheat cultivars Frontana and CSP44, respectively (Dyck et al. 1966; Bansal et al. 2008). The leaf rust resistance genes Lr13 and Lr23 have been used extensively in breeding programs in India, and most of the older cultivars and breeding lines carry these genes; presently, these genes are not effective. Moreover, the leaf rust resistance genes, Lr16 and Lr48 are not effective against all four pathotypes, and the reaction of these pathotypes to Lr73 is unknown. One QTN, Q.Lr.iari-2DS_12-5, detected on chromosome 2DS and associated with the 12-5 pathotype was found to be closely associated with the QLr.ags-2DS QTL (Sapkota et al. 2019a). Interestingly, QTN, Q.Lr.iari-3AL.1_77-5, was identified at 633.05 Mb on the long arm of chromosome 3A and was consistently associated with two pathotypes, i.e., 77-5 and 104-2, which is near the QYr.hebau-3AL/QLr.hebau-3AL QTL associated with multiple rust resistance (Gebrewahid et al. 2020).
Five QTNs, QLr.iari-3BL.1_77-5, QLr.iari-3BL.2_77-5, QLr.iari-3BL.3_77-5, QLr.iari-3BL_77-9 and QLr.iari-3BL_104-2, were found on the long arm of chromosome 3B located at 803.20-817.01 Mb, which is close to the QLr.hzau-3BL QTL (Zhou et al. 2021) and markers, IWA6633,IWA7889 and IWA4312 (Turner et al. 2017). For pathotype 77-5, QLr.iari-1AS_77-5 on the short arm of chromosome 1A was close to Lr10 (Choudhuri 1958), QLr.iari-1DL_77-5 was close to Lr21 (Rowland and Kerber 1974), and Lr38 (Friebe et al. 1993) was located on the long arm of chromosome 1D. Pathotypes, 77-5 and 77-9 are virulent on the leaf rust resistance genes, Lr10, Lr21 and Lr38. Seven other QTNs, i.e., QLr.iari-3DL.1_12-5, QLr.iari-3DL.2_12-5, QLr.iari-3DL_77-5, QLr.iari-3DL.1_77-9, QLr.iari-3DL.2_77-9, QLr.iari-3DL.1_104-2 and QLr.iari-3DL.2_104-2 were found on chromosome 3DL, which was close to the gene, Lr24 (Mclntosh et al. 1976). The leaf rust resistance gene, Lr24 has been providing resistance to leaf rust for more than two decades in India, so the presence of Lr24 is obvious in cultivars and advance breeding lines. Out of 7 QTNs, QLr.iari-3DL.1_12-5 and QLr.iari-3DL.1_77-9 were associated with marker AX-95226287 for two pathotypes, i.e., 12-5, 77-9. Similarly, QLr.iari-3DL.2_12-5 and QLr.iari-3DL.2_77-9 were consistent with the 12-5 and 77-9 pathotypes associated with the AX-95211729 marker. Two QTNs, QLr.iari-3DS.1_77-5 and QLr.iari-3DS.2_77-5, identified in pathotype 77-5 were located on chromosome 3DS near the genes, Lr32 (Kerber, 1987) and Lr38 (Friebe et al. 1993). On chromosome 6DL, QLr.iari-6DL_77-9 and QLr.iari-6DL_77-5 QTNs were close to Lr38 (Friebe et al. 1993). Pathotypes 77-5 and 77-9 are avirulent on Lr32 and virulent on Lr38. Similarly, ten QTNs were also detected closer to loci reported in previous studies, which were distributed on chromosomes 6BS (QLr.usw-6BS: Kthiri et al. 2019), 6DS (IWA2476: Turner et al. 2017), 7AL (IWA4175: Turner et al. 2017), 7AS (IWA1277: Turner et al. 2017), 7BL (QLr.usw-7BL: Kthiri et al. 2019) and 7DS (KaspLr34: Turner et al. 2017).
Furthermore, fifteen QTNs were consistently identified at the adult plant stage to LR in two or more models and were distributed on Chr1A, Chr1B, Chr1D, Chr2A, Chr2B, Chr3A, Chr3B, Chr3D, Chr4A, Chr4D and Chr6D, with phenotypic variation ranging between 1.30-17.83%. Out of 15 QTNs, one QTN QLr.iari-1AL_APR detected on chromosome 1AL is close to the QLr.ags-1AL QTL (Sapkota et al. 2019a) and the IWA1557 marker (Turner et al. 2017). Similarly, QTN QLr.iari-1BL_APR found on the long arm of chromosome 1B is close to QLr.hebau-1BL (Xu et al. 2021) and QLr.sun-1BL (Kandiah et al. 2020) QTLs. Another QTN, QLr.iari-2BL_APR, located at 596.41 Mb on chromosome 2BL is close to QLr.sun-2BL (Kandiah et al. 2020) and (IWA2025, IWA207, IWA2509: Turner et al. 2017). Interestingly, one chromosomal region, i.e., 3BL was associated with four QTNs: QLr.iari-3AL.1_APR, QLr.iari-3AL.2_APR, QLr.iari-3AL.3_APR and QLr.iari-3AL.4_APR are near QYr.hebau-3AL/QLr.hebau-3AL (Gebrewahid et al. 2020). Similarly, two QTNs were found near those reported in previous studies, which are distributed on chromosomes 3BL (IWA6633, IWA7889, IWA4312: Turner et al. 2017) and 4AL (QLr.hebau-4AL: Zhang et al. 2017b). Of these 15 QTNs, five were novel QTNs, i.e., 1DL: QLr.iari-1DL_APR, 2AL: QLr.iari-2AL_APR, 3DL: QLr.iari-3DL_APR, 4DL: QLr.iari-4DL_APR and 6DL, QLr.iari-6DL_APR, which were not reported in previous studies.
The identification and utilization of novel genes/QTNs for resistance is a continuous process and a regular challenge that is critical in plant breeding to cope with the threats to crop production caused by diseases. Genome-wide association studies are a powerful approach to detect QTNs associated with resistance. In this study, the significance of six ML-GWAS models on a panel of various wheat genotypes provided comprehensive insight into the molecular genetic basis of leaf rust resistance in wheat. A total of 51 reliable QTNs for seedling and 15 QTNs for APR were detected by two or more models. Of these QTNs, 8 QTNs showed a significant effect on the corresponding leaf rust pathotypes. The results of the current study showed the value of our diverse genetic resources conserved in the Indian National Gene bank. Although many QTNs were identified in the known region, the presence of novel QTN regions (i.e., 1BS, 3AS, 5AL, 5DL and 7BS) for four pathotypes at the seedling stage and (1DL, 2AL, 3DL, 4DL and 6DL) for the adult plant stage could enhance our understanding and provide new resources for leaf rust resistance in wheat. Furthermore, the novel genomic regions identified in the present study must be examined for favorable alleles associated with resistance genes so that they can be used in breeding programs.
Data availability
The original contributions presented in the study are included in the article/Supplementary Files. Genotype and phenotype data have been submitted to Dryad (https://doi.org/10.5061/dryad.h9w0vt4kq).
References
Afzal AJ, Wood AJ, Lightfoot DA (2008) Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol Plant–Microbe Interact 21:507–517
Aoun M, Breiland M, Turner MK, Loladze A, Chao SX, Xu SS et al. (2016) Genome-wide association mapping of leaf rust response in a durum wheat worldwide germplasm collection. Plant Genome 9:3
Aoun M, Kolmer JA, Rouse MN, Elias EM, Breiland M, Bulbula WD et al. (2019) Mapping of novel leaf rust and stem rust resistance genes in the Portuguese durum wheat landrace. PI 192051 G3 9:2535–2547
Atwell S, Huang YS, Vilhja BJ, Meng D, Platt A, Tarone AM et al. (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631. https://doi.org/10.1038/nature08800
Bansal UK, Hayden MJ, Venkata BP, Khanna R, Saini RG, Bariana HS (2008) Genetic mapping of adult plant leaf rust resistance genes Lr48 and Lr49 in common wheat. Theor Appl Genet 117:307–312. https://doi.org/10.1007/s00122-008-0775-6
Bhardwaj SC (2011) Resistance genes and adult plant rust resistance of released wheat varieties of India. Res Bull 5–31
Bhatia G, Upadhyay SK, Upadhyay A, Singh K (2021) Investigation of long noncoding RNAs as regulatory players of grapevine response to powdery and downy mildew infection. BMC Plant Biol 21:1–16
Bokore FE, Cuthbert RD, Knox RE, Randhawa HS, Hiebert CW, DePauw RM et al. (2017) Quantitative trait loci for resistance to stripe rust of wheat revealed using global field nurseries and opportunities for stacking resistance genes. Theor Appl Genet 130:2617–2635
Bolton MD, Kolmer JA, Garvin DF (2008) Wheat leaf rust caused by Pucciniatriticina. Mol Plant Pathol 9:563–575
Brachi B, Morris GP, Borevitz JO (2011) Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol 12:232
Bradbury J, Zhang Z, Kroon DE, Casstevens M, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Breseghello F, Sorrells FM (2005) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177. https://doi.org/10.1534/genetics.105.044586
Chao S, Dubcovsky J, Dvorak J, Luo MC, Baenziger SP, Matnyazov R, Clark DR, Talbert LE, Anderson JA, Dreisigacker S, Glover K (2010) Population- and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.). BMC Genomics 11:1–17
Choudhuri HC (1958) The inheritance of stem and leaf rust resistance in common wheat. Indian J Genet 18:90–115
Cleveland WS (1979) Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc 74:829–836. https://doi.org/10.1080/01621459.1979.10481038
Cloutier S, Wang Z, Banks TW, Jordan MC, McCallum BD (2008) Gene expression of plant defence pathways using Lr1 transgenic lines and the Affymetrix wheat chip
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Danakumara T, Kumari J, Singh AK, Sinha SK, Pradhan AK, Sharma S et al. (2021) Genetic dissection of seedling root system architectural traits in a diverse panel of hexaploid wheat through multilocus genome-wide association mapping for improving drought tolerance. Int J Mol Sci 22:7188
Dinkar V, Jha SK, Mallick N, Niranjana M, Agarwal P, Sharma JB (2020) Molecular mapping of a new recessive wheat leaf rust resistance gene originating from Triticum spelta. Sci Rep 10:1–9
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dyck PL, Samborski DJ, Anderson RG (1966) Inheritance of adult-plant leaf rust resistance derived from the common wheat varieties Exchange and Frontana. Can J Genet Cytol 8:665–671
Edae EA, Pumphrey MO, Rouse MN (2018) A genome-wide association study of field and seedling response to individual stem rust pathogen races reveals combinations of race-specific genes in North American spring wheat. Front Plant Sci 9:52
Ellis JG, Lagudah ES, Spielmeyer W, Dodds PN (2014) The past, present and future of breeding rust resistant wheat. Front Plant Sci 5:641
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Feuillet C, Schachermayr G, Keller B (1997) Molecular cloning of a new receptor‐like kinase gene encoded at the Lr10 disease resistance locus of wheat. Plant J 11:45–52
Flor HH (1956) The complementary genic systems in flax and flax rust. Adv Genet 8:29–54
Friebe B, Jiang J, Gill BS, Dyck PL (1993) Radiation-induced nonhomoeologous wheat-Agropyron intermedium chromosomal translocations conferring resistance to leaf rust. Theor Appl Genet 86:141–149
Gebrewahid TW, Zhang P, Zhou Y, Yan X, Xia X, He Z et al. (2020) QTL mapping of adult plant resistance to stripe rust and leaf rust in a Fuyu 3/Zhengzhou 5389 wheat population. Crop J 8:655–665
Gupta SK, Rai AK, Kanwar SS, Sharma TR (2012) Comparative analysis of zinc finger proteins involved in plant disease resistance. PLoS ONE 7:e42578
Hall D, Tegström C, Ingvarsson PK (2010) Using association mapping to dissect the genetic basis of complex traits in plants. Brief Funct Genomics Proteom 9:157–165
Huerta-Espino J, Singh R, German S, McCallum B, Park R, Chen WQ et al. (2011) Global status of wheat leaf rust caused by Pucciniatriticina. Euphytica 179:143–160
Jain N, Batra R, Saripalli G, Sinha N et al. (2021) Methylome changes during Lr48-mediated APR for leaf rust in wheat (Triticum aestivum L.). Physio Mol Plant Pathol 116:101726. https://doi.org/10.1016/j.pmpp.2021.101726
Jaiswal V, Mir RR, Mohan A, Balyan HS, Gupta PK (2012) Association mapping for preharvest sprouting tolerance in bread wheat (Triticum aestivum L.). Euphytica 188:89–102
Johnson R (1984) A critical analysis of durable resistance. Annu Rev Phytopathol 22:309–330
Joukhadar R, Daetwyler HD, Bansal UK, Gendall AR, Hayden MJ (2017) Genetic diversity, population structure and ancestral origin of Australian wheat. Front Plant Sci 8:2115
Joukhadar R, Hollaway G, Shi F, Kant S, Forrest K, Wong D et al. (2020) Genome- wide association reveals a complex architecture for rust resistance in 2300 worldwide bread wheat accessions screened under various Australian conditions. Theor Appl Genet 133:2695–2712. https://doi.org/10.1007/s00122-020-03626-9
Juliana P, Singh RP, Singh PK, Poland JA, Bergstrom GC, Huerta-Espino J et al. (2018) Genome-wide association mapping for resistance to leaf rust, stripe rust and tan spot in wheat reveals potential candidate genes. Theor Appl Genet 131:1405–1422
Kaler AS, Gillman JD, Beissinger T, Purcell LC (2020) Comparing different statistical models and multiple testing corrections for association mapping in soybean and maize. Front Plant Sci 10:1794
Kandiah P, Chhetri M, Hayden M, Ayliffe M, Bariana H, Bansal U (2020) Mapping of adult plant leaf rust resistance in Aus27506 and validation of underlying loci by in-planta fungal biomass accumulation. Agronomy 10:943
Kerber ER (1987) Resistance to leaf rust in hexaploid wheat: Lr32 a third gene derived from Triticum tauschii. Crop Sci 27:204–206. https://doi.org/10.2135/cropsci1987.0011183X002700020013x
Kertho A, Mamidi S, Bonman JM, McClean PE, Acevedo M (2015) Genome-wide association mapping for resistance to leaf and stripe rust in winter-habit hexaploid wheat landraces. PLoS One 10:e0129580
Kolmer J (2013) Leaf rust of wheat: pathogen biology, variation and host resistance. Forests 4:70–84
Kthiri D, Loladze A, N’Diaye A, Nilsen KT, Walkowiak S, Dreisigacker S et al. (2019) Mapping of genetic loci conferring resistance to leaf rust from three globally resistant durum wheat sources. Front plant Sci 10:1247
Kulwal PL, Mir RR, Varshney RK (2022). Efficient breeding of crop plants. In: Yadava D. K. et al. (eds.), Fundamentals of field crop breeding, https://doi.org/10.1007/978-981-16-9257-4_14
Kumar D, Chhokar V, Sheoran S, Singh R, Sharma P, Jaiswal S et al. (2020a) Characterization of genetic diversity and population structure in wheat using array based SNP markers. Mol Biol Rep 47:293–306
Kumar D, Sharma S, Sharma R, Pundir S, Singh VK, Chaturvedi D et al. (2021) Genome-wide association study in hexaploid wheat identifies novel genomic regions associated with resistance to root lesion nematode (Pratylenchusthornei). Sci Rep 11:1–14
Kumar S, Kumari J, Bhusal N, Pradhan AK, Budhlakoti N, Mishra DC et al. (2020b) Genome-wide association study reveals genomic regions associated with ten agronomic traits in wheat under late-sown conditions. Front Plant Sci 11:1420
Kumar S, Phogat BS, Vikas VK, Sharma AK, Saharan MS, Singh AK et al. (2019) Mining of Indian wheat germplasm collection for adult plant resistance to leaf rust. PloS One 14:e0213468
Levene H (1961). Robust tests for equality of variances. Contributions to probability and statistics. Essays in honor of Harold Hotelling, 279–292.
Li D, Liu H, Zhang H, Wang X, Song F (2008) OsBIRH1, a DEAD-box RNA helicase with functions in modulating defence responses against pathogen infection and oxidative stress. J Exp Bot 59:2133–2146
Li H, Wei C, Meng Y, Fan R, Zhao W, Wang X et al. (2020) Identification and expression analysis of some wheat F-box subfamilies during plant development and infection by Pucciniatriticina. Plant Physiol Biochem 155:535–548
Lipka AE, Gore MA, Magallanes-Lundback M, Mesberg A, Lin H, Tiede T et al. (2013) Genome-wide association study and pathway-level analysis of tocochromanol levels in maize grain. G3: Genes, Genomes Genet 3:1287–1299
Loegering WQ (1959) Methods for Recording Cereal Rust Data in International Spring Wheat Rust Nursery (IRN). USDA, Washington, DC
Marasas CN, Smale M, Singh RP (2004) The economic impact in developing countries of leaf rust resistance breeding in CIMMYT- related spring bread wheat. Economics Program Paper: 04-01.
Marone D, Russo MA, Laidò G, De Leonardis AM, Mastrangelo AM (2013) Plant nucleotide binding site–leucine-rich repeat (NBS-LRR) genes: active guardians in host defense responses. Int J Mol Sci 14:7302–7326
McCallum BD, Hiebert C, Huerta-Espino J, Cloutier S (2012) In: Sharma I, editor. Disease resistance in wheat. Wallingford: CAB International: 33–62.
McCartney CA, Somers DJ, McCallum BD, Thomas J, Humphreys G, Menzies JG et al. (2005) Microsatellite tagging of the leaf rust resistance gene Lr16 on wheat chromosome 2BSc. Mol Breed 15:329–337. https://doi.org/10.1007/s11032-004-5948-7
McDonald BA, Linde C (2002) The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica 124:163–180
McIntosh RA, Dubcovsky J, Rogers WJ, Morris C, Xia XC, Designators L (2017) Catalogue of gene symbols for wheat: 2017 supplement. Komugi Wheat Genet Resour database.
McIntosh RA, Dubcovsky J, Rogers WJ, Xia XC, Raupp WJ (2020) Catalogue of gene symbols for wheat: 2020 supplement. https://wheatpwusda.gov/GG3/wgc
McIntosh RA, Dyck PL (1975) Cytogenetical studies in wheat. VII Gene Lr23 for reaction to Puccinia recondita in Gabo and related cultivars. Aust. J Biol Sci 28:201–212. https://doi.org/10.1071/BI9750201
McIntosh RA, Dyck PL, Green GJ (1976) Inheritance of leaf rust and stem rust resistances in wheat cultivars Agent and Agatha. Aust J Agric Res 28:37–45
Mir RR, Zaman-Allah M, Sreenivasulu N, Trethowan R, Varshney RK (2012) Integrated genomics, physiology and breeding approaches for improving drought tolerance in crops. Theor Appl Genet 125:625–645
Mir RR, Kumar S, Shafi S (2021). Genetic dissection for yield and yield-related traits in bread wheat (Triticum aestivum L.). In: Wani SH, Mohan A, Singh GP (Eds.) Physiological, molecular, and genetic perspectives of wheat improvement, 209–227.
Mujeeb-Kazi A, Kazi AG, Dundas I, Rasheed A, Ogbonnaya F, Kishii M et al. (2013) Genetic diversity for wheat improvement as a conduit to food security. Adv Agron 4:179–257
Muleta KT, Bulli P, Rynearson S, Chen X, Pumphrey M (2017b) Loci associated with resistance to stripe rust (Pucciniastriiformisf. sp. tritici) in a core collection of spring wheat (Triticumaestivum). Plos One 12:e0179087
Muleta KT, Rouse MN, Rynearson S, Chen X, Buta BG, Pumphrey MO (2017a) Characterization of molecular diversity and genome-wide mapping of loci associated with resistance to stripe rust and stem rust in Ethiopian bread wheat accessions. BMC Plant Biol 17:134
Naoumkina MA, Zhao Q, Gallego-Giraldo L, Dai X, Zhao PX, Dixon RA (2010) Genomewide analysis of phenylpropanoid defence pathways. Mol Plant Pathol 11:829–846
Narayanan NN, Grosic S, Tasma IM, Grant D, Shoemaker R, Bhattacharyya MK (2009) Identification of candidate signaling genes including regulators of chromosome condensation 1 protein family differentially expressed in the soybean–Phytophthorasojae interaction. Theor Appl Genet 118:399
Nazeem PA, Jose S, Sheeba NK, Madhavan S, Baby A, Kumar PS, Devi N (2011) Differential gene expression for bacterial wilt incidence in tomato (Solanum lycopersicum L.) revealed by cDNA-AFLP analysis. Physiol Mol plant Pathol 76:197–203
Pariyar SR, Dababat AA, Sannemann W, Erginbas-Orakci G, Elashry A, Siddique S et al. (2016) Genome-wide association study in wheat identifies resistance to the cereal cyst nematode Heteroderafilipjevi. Phytopathology 106:1128–1138
Park RF, Mohler V, Nazari K, Singh D (2014) Characterization and mapping of Lr73 gene conferring seedling resistance to Puccinia triticina in common wheat. Theor Appl Genet 127:2041–2049. https://doi.org/10.1007/s00122-014-2359-y
Peterson RF, Campbell AB, Hannah AE (1948) A diagrammatic scale for estimating rust intensity on leaves and stems of cereals. Can J Res 26c:496–500
Pradhan AK, Kumar S, Singh AK, Budhlakoti N, Mishra DC, Chauhan D et al. (2020) Identification of QTLs/defense genes effective at seedling stage against prevailing races of wheat stripe rust in India. Front Genet, 11.
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Rakitsch B, Lippert C, Stegle O, Borgwardt K (2013) A Lasso multimarker mixed model for association mapping with population structure correction. Bioinformatics 29:206–214
Reif JC, Zhang P, Dreisigacker S, Warburton ML, Van Ginkel M, Hoisington D et al. (2005) Trends in genetic diversity during the history of wheat domestication and breeding. Theor Appl Genet 110:859–864
Riaz A, Athiyannan N, Periyannan SK, Afanasenko O, Mitrofanova OP, Platz GJ (2018) Unlocking new alleles for leaf rust resistance in the Vavilov wheat collection. Theor Appl Genet 131:127–144
Riaz A, Periyannan S, Aitken E, Hickey L (2016) A rapid phenotyping method for adult plant resistance to leaf rust in wheat. Plant Methods 12:17
Riedelsheimer C, Lisec J, Czedik-Eysenberg A, Sulpice R, Flis A, Grieder C et al. (2012) Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc Natl Acad Sci USA 109:8872–8877
Roelfs AP, Singh RP, Saari EE (1992) Rust Diseases of Wheat: Concepts and Methods of Disease Management. CIMMYT, Mexico
Rosewarne GM, Singh RP, Huerta-Espino J, Herrera-Foessel SA, Forrest KL, Hayden MJ et al. (2012) Analysis of leaf and stripe rust severities reveals pathotype changes and multiple minor QTLs associated with resistance in an Avocet × Pastor wheat population. Theor Appl Genet 124:1283–1294. https://doi.org/10.1007/s00122-012-1786-x
Rowland GG, Kerber ER (1974) Telocentric mapping in hexaploid wheat of genes for leaf rust resistance and other characters derived from Aegilops squarrosa. Can J Genet Cytol 16:137–144
Saintenac C, Zhang W, Salcedo A, Rouse MN, Trick HN, Akhunov E et al. (2013) Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341:783–786
Sapkota S, Hao Y, Johnson J, Buck J, Aoun M, Mergoum M (2019a) Genome wide association study of a worldwide collection of wheat genotypes reveals novel quantitative trait loci for leaf rust resistance. The Plant. Genome 12:190033
Sapkota S, Hao YF, Johnson J, Lopez B, Bland D, Chen ZB et al. (2019b) Genetic mapping of a major gene for leaf rust resistance in soft red winter wheat cultivar AGS 2000. Mol Breed 39:8
Saripalli G, Singh K, Gautam T, Kumar S, Raghuvanshi S, Prasad P et al. (2020) Genome-wide analysis of H3K4me3 and H3K27me3 modifications due to Lr28 for leaf rust resistance in bread wheat (Triticumaestivum). Plant Mol Biol 104:113–136
SAS Institute Inc. (2017) SAS/STAT® 14.3 User’s Guide: High-Performance Procedures. Cary, NC: SAS Institute Inc
Sauvage C, Segura V, Bauchet G, Stevens R, Do PT, Nikoloski Z et al. (2014) Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol 165:1120–1132
Segura V, Vilhjálmsson BJ, Platt A, Korte A, Seren Ü, Long Q et al. (2012) An efficient multilocus mixed-model approach for genome wide association studies in structured populations. Nat Genet 44:825–830. https://doi.org/10.1038/ng.2314
Semagn K, Bjørnstad Å, Xu Y (2010) The genetic dissection of quantitative traits in crops. Electron J Biotechnol 13:16–17
Serba DD, Uppalapati SR, Mukherjee S, Krom N, Tang Y, Mysore KS et al. (2015) Transcriptome profiling of rust resistance in switchgrass using RNA‐Seq analysis. Plant Genome 8:2014–10
Sharma C, Kumar S, Saripalli G, Jain N, Raghuvanshi S, Sharma JB et al. (2019) H3K4/K9 acetylation and Lr28-mediated expression of six leaf rust responsive genes in wheat (Triticumaestivum). Mol Genet Genom 294:227–241
Sheoran S, Jaiswal S, Kumar D, Raghav D, Sharma N, Pawar R et al. (2019) Uncovering genomic regions associated with 36 agro-morphological traits in Indian spring wheat using GWAS. Front Plant Sci 10:527
Shiferaw B, Smale M, Braun H-J, Duveiller E, Reynolds M, Muricho G (2013) Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security Food Sec 5:291–317. https://doi.org/10.1007/s12571-013-0263-y
Singh D, Kumar D, Satapathy L, Pathak J, Chandra S, Riaz A et al. (2017) Insights of Lr28 mediated wheat leaf rust resistance: Transcriptomic approach. Gene 637:72–89
Snedecor GW, Cochran WG (1989) Statistical methods 8th ed., (235-236).
Sonawala U, Dinkeloo K, Danna CH, McDowell JM, Pilot G (2018) Functional linkages between amino acid transporters and plant responses to pathogens. Plant Sci 277:79–88
Song X, Rampitsch C, Soltani B, Mauthe W, Linning R, Banks T et al. (2011) Proteome analysis of wheat leaf rust fungus, Puccinia triticina, infection structures enriched for haustoria. Proteomics 11:944–963
Stakman EC, Stewart DM, Loegering WQ (1962) Identification of Physiologic Races of Puccinia graminis var. Tritici. USDA, Washington, DC
Sukumaran S, Reynolds MP, Sansaloni C (2018) Genome-wide association analyses identify QTL hotspots for yield and component traits in durum wheat grown under yield potential, drought, and heat stress environments. Front Plant Sci 9:81
Tamba CL, Ni YL, Zhang YM (2017) Iterative sure independence screening EM-Bayesian LASSO algorithm for multilocus genome-wide association studies. PLoS computational Biol 13:e1005357
Tamba CL, Zhang YM (2018) A fast mrMLM algorithm for multilocus genome-wide association studies. Biorxiv, 341784
Turner MK, Kolmer JA, Pumphrey MO, Bulli P, Chao S, Anderson JA (2017) Association mapping of leaf rust resistance loci in a spring wheat core collection. Theor Appl Genet 130:345–361
Vaughn JN, Nelson RL, Song Q, Cregan PB, Li Z (2014) The genetic architecture of seed composition in soybean is refined by genome-wide association scans across multiple populations. G3: Genes, Genomes, Genet 4:2283–2294
Voss‐Fels K, Frisch M, Qian L, Kontowski S, Friedt W, Gottwald S, Snowdon RJ (2015) Subgenomic diversity patterns caused by directional selection in bread wheat gene pools. Plant Genome 8:2015–03
Wang SB, Feng JY, Ren WL, Huang B, Zhou L, Wen YJ et al. (2016a) Improving power and accuracy of genome-wide association studies via a multilocus mixed linear model methodology. Sci Rep. 6:1–10
Wang CF, Yin GH, Xia XC, He ZH, Zhang PP, Yao ZJ et al. (2016b) Molecular mapping of a new temperature-sensitive gene LrZH22 for leaf rust resistance in Chinese wheat cultivar Zhoumai 22. Mol Breed 36:18. https://doi.org/10.1007/s11032-016-0437-3
Wen YJ, Zhang H, Ni YL, Huang B, Zhang J, Feng JY (2018) Methodological implementation of mixed linear models in multilocus genome-wide association studies. Brief Bioinforma 19:700–712
Wu J, Gao J, Bi W, Zhao J, Yu X, Li Z et al. (2019) Genome-wide expression profiling of genes associated with Lr47-mediated wheat resistance to leaf rust (Puccinia triticina). Int J Mol Sci 20:4498
Xu X, Duan Z, Li X, Wu J, Yao Z (2021) QTL mapping of adult-plant resistance to leaf rust based on SSR markers and SNP sequencing of Chinese wheat landrace Xu’ai (Triticum aestivum L.). Genet Resour Crop Evol 68:1359–1373
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Zanke CD, Rodemann B, Ling J, Muqaddasi QH, Plieske J, Polley A et al. (2017) Genome-wide association mapping of resistance to eyespot disease (Pseudocercosporella herpotrichoides) in European winter wheat (Triticum aestivum L.) and fine-mapping of Pch1. Theor Appl Genet 130:505–514
Zhang G, Sun YF, Li YM, Dong YL, Huang XL, Yu YT et al. (2013) Characterization of a wheat C2 domain protein encoding gene regulated by stripe rust and abiotic stresses. Biol Plant 57:701–710
Zhang J, Feng JY, Ni YL, Wen YJ, Niu Y, Tamba CL et al. (2017a) pLARmEB: integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 118:517–524
Zhang P, Yin G, Zhou Y, Qi A, Gao F, Xia X et al. (2017b) QTL mapping of adult-plant resistance to leaf rust in the wheat cross Zhou 8425B/Chinese Spring using high-density SNP markers. Front plant Sci 8:793
Zhang Y-M, Jia Z, Dunwell JM (2019) The applications of new multilocus GWAS methodologies in the genetic dissection of complex traits. Front Plant Sci 2019:100
Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH et al. (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryzasativa. Nat Commun 2:467
Zhou Y, Tang H, Cheng MP, Dankwa KO, Chen ZX, Li ZY et al. (2017) Genome-wide association study for preharvest sprouting resistance in a large germplasm collection of chinese wheat landraces. Front Plant Sci 8:401
Zhou, J, Singh RP, Ren Y, Bai B, Li Z, Yuan C et al. (2021) Identification of two new loci for adult plant resistance to leaf rust and stripe rust in the Chinese wheat variety ‘Neimai 836’. Plant Dis.
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Gen 1:5–20
Ziems LA, Hickey LT, Hunt CH, Mace ES, Platz GJ, Franckowiak JD et al. (2014) Association mapping of resistance to Puccinia hordei in Australian barley breeding germplasm. Theor Appl Genet 127:1199–1212
Acknowledgements
The authors are highly thankful to the Indian Council of Agricultural Research for the financial support provided for this work. The authors are also thankful to the National Bureau of Plant Genetic Resources, New Delhi for providing germplasm lines for evaluation; the Indian Agricultural Statistics Research Institute, New Delhi for providing computational support; and the Indian Agricultural Research Institute, Regional Station, Wellington and Indian Institute of Wheat and Barley Research, Karnal for providing all support for screening wheat genotypes for leaf rust. The Authors duly acknowledge efforts of Dr. Philomin Juliana, Associate scientist, CIMMYT for English language editing.
Funding
Financial assistance from ICAR- National Bureau of Plant Genetic Resources through ICAR, CABin project Identification of novel QTLs/defence genes against rust in wheat for India (Project code-1006445).
Author information
Authors and Affiliations
Contributions
VKV: Conceptualization, data curation, data analysis, investigation, phenotype data of adult plant stage, writing-review and editing. AKP: Conceptualization, data curation, data analysis and data interpretation, investigation, writing-review and editing. NB: Conceptualization, data analysis, writing-review and editing. DCM: Contributed to the bioinformatics analysis. TC: Formal analysis and writing respective sections. SCB: Phenotypic evaluation of four different pathotypes of leaf rust at seedling stage and editing manuscript. SK: Phenotypic evaluation of four different pathotypes of leaf rust at seedling stage. MS: Provided phenotypic data for leaf rust resistance at the adult plant stage. PJ: Provided phenotypic data for leaf rust resistance at adult plant stage. MG: Provided phenotypic data for leaf rust resistance at adult plant stage. RN: Provided phenotypic data for leaf rust resistance at the adult plant stage. PS: Provided phenotypic data for leaf rust resistance at the adult plant stage. JP: Provided phenotypic data for leaf rust resistance at the adult plant stage. RRM: Writing-review and editing. KS: Writing-review and editing. Sundeep Kumar: Conceptualization, data curation, data analysis, investigation, writing-review and editing. All authors contributed to the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Lindsey Compton.
Rights and permissions
About this article

Cite this article
Vikas, V.K., Pradhan, A.K., Budhlakoti, N. et al. Multi-locus genome-wide association studies (ML-GWAS) reveal novel genomic regions associated with seedling and adult plant stage leaf rust resistance in bread wheat (Triticum aestivum L.). Heredity 128, 434–449 (2022). https://doi.org/10.1038/s41437-022-00525-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00525-1
This article is cited by
-
Exploring genomic regions involved in bread wheat resistance to leaf rust at seedling/adult stages by using GWAS analysis
BMC Genomics (2023)
-
Multi-faceted approaches for breeding nutrient-dense, disease-resistant, and climate-resilient crop varieties for food and nutritional security
Heredity (2022)
