
Abstract
Meiosis is undoubtedly the mechanism that underpins Mendelian genetics. Meiosis is a specialised, reductional cell division which generates haploid gametes (reproductive cells) carrying a single chromosome complement from diploid progenitor cells harbouring two chromosome sets. Through this process, the hereditary material is shuffled and distributed into haploid gametes such that upon fertilisation, when two haploid gametes fuse, diploidy is restored in the zygote. During meiosis the transient physical connection of two homologous chromosomes (one originally inherited from each parent) each consisting of two sister chromatids and their subsequent segregation into four meiotic products (gametes), is what enables genetic marker assortment forming the core of Mendelian laws. The initiating events of meiotic recombination are DNA double-strand breaks (DSBs) which need to be repaired in a certain way to enable the homologous chromosomes to find each other. This is achieved by DSB ends searching for homologous repair templates and invading them. Ultimately, the repair of meiotic DSBs by homologous recombination physically connects homologous chromosomes through crossovers. These physical connections provided by crossovers enable faithful chromosome segregation. That being said, the DSB repair mechanism integral to meiotic recombination also produces genetic transmission distortions which manifest as postmeiotic segregation events and gene conversions. These processes are non-reciprocal genetic exchanges and thus non-Mendelian.
Similar content being viewed by others

Introduction
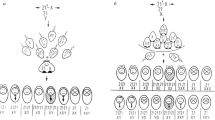
Meiosis is a specialised type of cell division that results in the production of gametes (reproductive cells). During meiosis diploid progenitor cells undergo two sequential rounds of division without an intervening round of DNA replication. As a consequence, the hereditary material is halved to produce gametes, which are haploid meiotic products. Gametes in turn fuse during fertilisation forming a zygote which, like the parents, is diploid. During meiosis, chromosomes are also re-assorted and recombined, so that the gametes formed contain new chromosome configurations. The independent assortment of whole chromosomes already generates genetic diversity in the gametes. However, to shuffle the hereditary material within a chromosome, deliberate breakage of chromosomes (DNA double-strand breaks, DSBs) by the DNA topoisomerase-II-related transesterase Spo11 and subsequent repair into new combinations is required (Lam and Keeney 2015; Zickler and Kleckner 2015; Hunter 2015) (Figs. 1 and 2). To halve the hereditary material accurately, homologous chromosomes (homologues) must be segregated faithfully from each other in the first meiotic division. Damaging the hereditary material through the formation of DSBs is potentially hazardous. However, inducing multiple DSBs on each chromosome allows the homologues to find each other when DSB ends start searching for homologous repair templates (in invertebrates initial homologue recognition and pairing is protein-mediated and independent of DSBs) (Zickler and Kleckner 2015). In the end, the DSB repair by meiotic homologous recombination physically connects the homologues via crossovers. These physical connections enable faithful chromosome segregation. Since a single crossover connecting each homologue pair is sufficient for guiding correct chromosome segregation (Kan et al. 2011), the majority of DSBs are actually repaired as non-crossovers using the homologue as a template, or redirected for repair using the sister chromatid. Moreover, the process of meiotic homologous recombination is important for evolution, because it increases the genetic diversity in populations. This in turn promotes the fitness of natural populations, since it enables various features of parents to be distributed to their progeny in novel combinations. Indeed, meiotic chromosome segregation and meiotic recombination are at the core of what we call the Mendelian laws of heredity (Fig. 1). These two processes mechanistically enable the law of segregation and the law of independent assortment, and they also underpin Thomas Hunt Morgan’s chromosomal theory of inheritance (Bateson 1909; Morgan 1910). However, the repair of DSBs during meiosis also leads to outcomes which are non-Mendelian in nature: postmeiotic segregation and gene conversion (Fig. 1). (Please note, that gene conversion is not restricted to meiosis, it can also occur in vegetative cells during homology-directed DNA repair. This, however, will not generate genetic transmission distortions in reproductive cells). In this review, we discuss how these non-Mendelian events were discovered and what we understand about the molecular processes generating them.

Recombination performed nearby such a heterozygous site will generate heteroduplex DNA (hDNA) containing mismatches which can lead to non-Mendelian segregation events.

A homologous chromosome pair is represented by blue and red sister chromatids. For clarity only the chromatids involved in recombination are shown, except for the initial step of the pathways and the final step of canonical Synthesis-Dependent Strand Annealing (SDSA). Please, note that the small gap representing the DSB does not indicate loss of genetic material as with Spo11 double cutting (see Fig. 3). Early steps of recombination are shown: DSB formation by Spo11, DNA strand resection to expose 3’ single-stranded tails which then invade a homologous template to form Displacement loops (D-loops). D-loops can be dissociated (by the action of DNA helicases; reviewed in Lorenz 2017) before or after DNA synthesis has started; antirecombination driven by MutSα, MutSβ, and MutLα also plays a key role here (see main text). Canonical SDSA is thought to produce non-crossover gene conversion events. The position of the initiating DSB site is indicated by a green vertical line. Dependent on the actions of mismatch repair a given heterozygous site within hDNA can be left unrepaired (PMS, 5:3 segregation), converted (gene conversion, 6:2 segregation), or restored (Mendelian 4:4 segregation) (see main text and Fig. 1 for details). Multiple invasion/dissociation cycles can result in complex conversion events when template switches between sister chromatids and homologues occur. The simultaneous or consecutive invasion of both ends of the DSB into the same or different chromatids of the homologue can result in double SDSA, were conversion tracts left and right of the original DSB site can be detected.
According to the ‘Glossary of Genetics’ (Rieger et al. 1991), gene conversion is “the nonreciprocal recombinational transfer of genetic information between homologous DNA sequences (allelic or homologous nonallelic genes) without an accompanying information exchange.” Indeed, the term “Genkonversion” (German for gene conversion) was coined by Hans Winkler in 1930 to explain meiotic recombination outcomes in fungi (Brunswik 1926) and mosses (von Wettstein 1924), which were incongruous with crossing over events (Winkler 1930; Lindegren 1949, 1958). Presumably, because Winkler originally pitted his gene-conversion theory directly against Morgan’s crossing-over theory (Morgan 1910), and because the main discerning feature of a gene conversion was that it could not be explained as a crossover (Winkler 1930; Lindegren 1949, 1958; Rieger et al. 1991), some authors use the terms gene conversion and non-crossover synonymously; this is particularly an issue in the older literature. However, a few decades after the publication of Winkler’s gene-conversion theory (Winkler 1930), further work in the ascomycetes Neurospora and Saccharomyces established that gene conversions can also be associated with crossovers, and that these recombination outcomes are mechanistically linked rather than mutually exclusive (Case and Giles 1958; Whitehouse and Hastings 1965; Fogel and Hurst 1967); this is now widely accepted (Fogel et al. 1979; Szostak et al. 1983). Much of what we understand about the molecular mechanisms underpinning the formation of gene conversion, comes from research in the two model yeast species Saccharomyces cerevisiae and Schizosaccharomyces pombe (see below). Although both species are unicellular fungi (yeasts), they are actually not closely related to each other, as they had their last common ancestor roughly at the same time as nematodes and mammals had theirs (Heckman et al. 2001). This already strongly indicates that gene conversion is a highly conserved process. Indeed, gene conversion has also been described in several multicellular eukaryotic species, including Drosophila melanogaster (e.g. Miller et al. 2012; Comeron et al. 2012), Arabidopsis thaliana (e.g. Sun et al. 2012; Drouaud et al. 2013; Wijnker et al. 2013), mouse (e.g. Cole et al. 2010, 2014; Gergelits et al. 2021), and human (e.g. Reiter et al. 1998; Jeffreys and May 2004). Caenorhabditis elegans is the only notable model for meiosis research, in which gene conversion between homologous chromosomes during meiotic recombination has not been demonstrated. However, gene conversion between homologues is likely to occur, as it does happen in the C. elegans germline during transposon excision (Robert et al. 2008) and between sister chromatids (Almanzar et al. 2021; Toraason et al. 2021).
After their formation by Spo11, meiotic DSBs undergo 5’→3’ resection of the DSB ends on one DNA strand to expose the other DNA strand as a 3’ single-stranded tail, which then invades homologous DNA sequences to mend the broken chromosome (Fig. 2) (Cejka and Symington 2021). There can be DNA sequence differences between homologous chromosomes, especially in natural populations where homologues are rarely identical. These DNA sequence differences will create mismatches within the recombination intermediates produced by the strand invasion process, so called heteroduplex DNA (hDNA) (Surtees et al. 2004; Spies and Fishel 2015) (Figs. 1 and 2). The fate of hDNA determines genetic recombination outcome essentially in three ways (Surtees et al. 2004; Spies and Fishel 2015). Firstly, if hDNA is left untouched the result is postmeiotic segregation (PMS), this is seen as a 5:3 segregation of the involved DNA sequence differences in the progeny (Figs. 1 and 2). Secondly, if hDNA is mismatch-corrected towards the information on the invading strand (donor) the result is gene conversion (6:2 segregation) (Figs. 1 and 2). Thirdly, if the mismatch is corrected using the information on the invaded strand (acceptor) then a 4:4 segregation to the parental situation is restored (Figs. 1 and 2). PMS is rare in wild-type crosses of budding yeast and fission yeast, but happens at a much higher frequency in the absence of functional mismatch repair (Schär and Kohli 1993; Alani et al. 1994; Hunter and Borts 1997; Schär et al. 1997).
It has been suggested that full gene conversions can be generated independent of mismatch repair through the formation and repair of gaps at the DSB site (e.g. Szostak et al. 1983; Martini et al. 2011). Indeed, in mitotic cells the 3’ single-stranded DNA tail produced during DNA resection is unstable, and its shortening results in gapped DSBs (Zierhut and Diffley 2008). It is unclear whether this also occurs at Spo11-generated meiotic DSBs. However, it has recently been demonstrated that the formation of two meiotic DSBs by Spo11 on the same chromosome in close proximity (‘double cuts’), will generate gaps of ~30 – ~100 base pairs in size (Fig. 3). Such events are apparently quite common (1/5th of all DSB events), and their repair can directly result in 6:2 and 2:6 conversions (Fig. 3) (Johnson et al. 2021; Prieler et al. 2021).

For the sake of simplicity only a single sister chromatid (one in blue, one in red) per homologous chromosome is shown, except for the last step to illustrate the segregation patterns of the recombination outcome. The positions of the initiating DSB sites are indicated by green vertical lines.
Going forward, we will focus on gene conversion as the main non-Mendelian outcome of meiotic recombination in a wild-type setting.
How do we detect and measure non-Mendelian segregation events?
Measuring gene conversion frequency at single nucleotide polymorphisms, small confined marker genes, or genetically engineered marker constructs, depends on several factors. Gene conversions tend to be infrequent events, which makes their detection difficult. This is especially the case, when one parent contains a wild-type marker gene and the other parent has a single DNA change. Here, gene conversion can only be detected in so-called tetrad dissection experiments. Tetrad dissection enables the analysis of all 4 products of a single meiosis revealing the aberrant 3:1 or 1:3 (6:2 or 2:6) segregation patterns of wild-type vs. mutant marker genes (Winkler 1930; Lindegren 1958). Dissecting hundreds, if not thousands, of tetrads to observe a few gene conversion events is laborious. If half-chromatids need to be studied to discern gene conversion from PMS events, this becomes even more involved. There are two biological features exploited in the last few decades which alleviated some of the challenges around measuring gene conversion frequency at specific markers. One is the discovery of so-called meiotic recombination hotspots (simply referred to as hotspots from here onwards), and the other is the use of 2-point or bifactorial crosses.
Hotspots are DNA sites or regions with a higher-than-average frequency of meiotic recombination (Petes 2001; Wahls and Davidson 2012). In the two model yeast species, there are natural hotspots (e.g. Lichten and Goldman 1995; Cromie et al. 2005; Steiner and Smith 2005a), hotspots generated by point mutations (e.g. Ponticelli et al. 1988; Schuchert and Kohli 1988; Kon et al. 1997), and biotechnologically engineered hotspots (e.g. White and Petes 1994; Baur et al. 2005). The frequency of recombination events, including gene conversions, at hotspots is largely defined by the amount of DSBs made by Spo11 within the hotspot region (Petes 2001; Lam and Keeney 2015). Interestingly, it has also been shown in S. cerevisiae that hypomorphic mutants of SPO11 can affect gene conversion tract length differentially between non-crossover and crossover outcomes (Rockmill et al. 2013). This indicates that the extent of conversion at a specific locus can be misestimated, especially when it only contains a few scorable markers. Generally, the frequency of gene conversion will be higher at sites which receive large numbers of DSBs; this then requires fewer tetrad dissections to obtain meaningful data.
The second technical improvement is looking at 2-point or bifactorial crosses. Rather than studying gene conversion in crosses where one parent carries a mutant allele and the other a wild-type allele (1-point or monofactorial crosses), in bifactorial crosses parents with two different alleles (heteroalleles) at the same test locus are crossed with each other. Bifactorial crosses have a distinct advantage in that since both parents are mutant, only progeny having undergone gene conversion can be wild-type for the associated phenotype. Therefore, this circumvents the need to perform tetrad dissection to identify gene conversion events unambiguously (e.g. Gutz 1971; Schär et al. 1993; Zahn-Zabal and Kohli 1996). One pair of genes involved in adenine metabolism in both model yeasts, proved particularly useful for measuring gene conversion frequency in bifactorial crosses. These are ADE1 (coding for the SAICAR synthetase) and ADE2 (coding for the AIR carboxylase) in S. cerevisiae, and their orthologues in Sz. pombe, ade7+ and ade6+, respectively (Juang et al. 1993; Rébora et al. 2001). Mutations in these genes produce yeast colonies displaying a pink to red colour when grown under specific conditions. This makes it very easy to phenotype progeny which have undergone gene conversion restoring the wild-type creamy white colony colour, from bifactorial crosses where both parents carry mutant heteroalleles of these genes and thus form pink or red colonies (Lindegren 1949; Leupold 1958).
In Sz. pombe, identification of the M26 mutation in ade6 as a meiotic recombination and DSB hotspot (Schuchert and Kohli 1988; Steiner et al. 2002) enabled recombination frequency analysis at a hotspot in bifactorial crosses. Further improvements, including screening for hotter versions of the ade6-M26 allele (Steiner and Smith 2005b), and generating a genetic interval with scorable marker genes around ade6 to measure crossover frequency associated with gene conversion (Osman et al. 2003; Lorenz et al. 2010), make this a genetic tool used to this day.
Progress with high-density DNA microarray and whole-genome sequencing technologies also facilitated the mapping of recombination events in S. cerevisiae on a genome-wide scale. Here, 2 different strains of S. cerevisiae which harbour thousands of different natural genetic variants (single nucleotide polymorphisms, and small insertions & deletions) are crossed to each other, and the shuffled distribution of the genetic variants in the progeny of such hybrid meioses can be used to analyse gene conversion and PMS, as well as crossover and non-crossover frequencies (e.g. Chen et al. 2008; Mancera et al. 2008, 2011; Martini et al. 2011; Rockmill et al. 2013; Oke et al. 2014; Marsolier-Kergoat et al. 2018; Cooper et al. 2021; Johnson et al. 2021). Up to 1% of the genome can be subjected to gene conversion in each meiotic product (Mancera et al. 2008), indicating that the contribution of meiotic gene conversion to genetic diversity in progeny is substantial, but tends to be underestimated in comparison to crossovers (Cole et al. 2012).
The mismatch repair pathway governed by the MutSα (Msh2-Msh6), Mutsβ (Msh2-Msh3), and MutLα (Mlh1-Pms1) complexes is a major determinant of meiotic recombination outcome (Surtees et al. 2004; Spies and Fishel 2015). These particular MutS and MutL complexes influence meiotic recombination outcome on two levels (Surtees et al. 2004; Spies and Fishel 2015). Firstly, they repair mismatches in hDNA, thus driving restoration and conversion (Fig. 2). Secondly, MutS coordinates disassembly of recombination intermediates containing (too many) mismatches to ensure that repair is performed from a homologous template; this is called antirecombination. Mutants inactivating these functions of MutSα and MutLα thus serve as tools to enable the detection of hDNA and measure the length of DNA tracts in non-Mendelian segregation events. Notably, S. cerevisiae msh2∆ mutants have been exploited to improve our understanding of meiotic recombination mechanisms in an engineered polymorphic region harbouring a hotspot (Ahuja et al. 2021) and on a genome-wide scale (Martini et al. 2011; Marsolier-Kergoat et al. 2018; Cooper et al. 2021). This approach uncovered molecular details of non-Mendelian events during meiotic recombination at an unprecedented resolution (see below), but this also has its limitations because deletion of mutSα (and mutLα) genes does also affect overall gene conversion and crossover frequency (Martini et al. 2011; Brown et al. 2019; Ahuja et al. 2021; Cooper et al. 2021).
How do we interpret the genetic and molecular evidence?
hDNA tracts which subsequently manifest as PMS and gene conversion events can be associated with crossovers and non-crossovers (see above). All these events are initiated by programmed DSBs, but at which point in their further processing do the repair pathways leading to either crossovers or non-crossovers diverge? Answering this question will enable a detailed understanding of how given recombination outcomes are generated, which has profound implications for interpreting genetic data. Early models of meiotic recombination argued that crossovers and non-crossovers are produced by very different DSB repair pathways. Non-crossovers were considered to be the results of synthesis-dependent strand annealing (Fig. 2) (Resnick 1976), and both crossovers and non-crossovers were thought to be produced by the “DSB repair” pathway involving the formation of double Holliday junctions (Szostak et al. 1983). The name of the latter repair pathway is not ideal but has historical reasons. In these early models the resolution of Holliday junctions was proposed to be unbiased because these recombination intermediates are symmetric and would thus be processed into crossovers and non-crossovers at an equal rate. Over the years, these models were refined, as there were strands of evidence supporting an early divergence of non-crossover and crossover formation. Firstly, in various species the gene conversion tracts of crossovers are longer than that of non-crossovers (Mancera et al. 2008; Wijnker et al. 2013; Cole et al. 2014). Secondly, in S. cerevisiae non-crossover recombination intermediates arise earlier during meiosis than crossover ones (Allers and Lichten 2001). This led to the idea that non-crossovers are predominantly the result of synthesis-dependent strand annealing, whereas biased resolution of double Holliday junctions exclusively produces crossovers.
More recent experiments performed in S. cerevisiae revealed that non-Mendelian segregation tracts associated with crossovers and non-crossovers are considerably more complex than originally appreciated, but also more similar to one another (Figs. 2 and 4) (Martini et al. 2011; Oke et al. 2014; Marsolier-Kergoat et al. 2018; Ahuja et al. 2021). Also, in Drosophila melanogaster, conversion tracts in crossovers and non-crossovers are of similar length (Comeron et al. 2012), and display discontinuities in a Msh6 mutant (Radford et al. 2007). More importantly, many of the observed complexities in the hDNA are incompatible with non-crossovers and crossovers being generated by the simple models of synthesis-dependent strand annealing and DSB repair involving Holliday junctions, respectively. A unified hypothesis, the Disassembly/Migration-Annealing (D/M-A) model, addresses these issues (Lao et al. 2008; Ahuja et al. 2021). This D/M-A model basically suggests that both crossovers and non-crossovers are formed by synthesis-dependent strand annealing, which generates non-Mendelian segregation tracts (hDNA) in the process, and only subsequent events stabilise certain recombination intermediates to create double Holliday junctions (Ahuja et al. 2021). The resolution of these double Holliday junctions then predominantly, if not exclusively, results in crossovers (Fig. 4).

A homologous chromosome pair is represented by blue and red sister chromatids. For clarity only the chromatids involved in recombination are shown after the initial step of the pathways. Please, note that the small gap representing the DSB does not indicate loss of genetic material as with Spo11 double cutting (see Fig. 3). The early steps of recombination are shown as before (see Fig. 2). These include: DSB formation by Spo11, 5’ to 3’ resection of DSB ends on the broken DNA duplex (blue) to expose 3’ single-stranded tails, and strand invasion of the intact DNA duplex (red) to form a Displacement loop (D-loop). For crossover repair pathways, D-loop formation is then followed by DNA synthesis, helicase-mediated branch migration (grey arrows), capture of the second end of the DSB, further processing to produce double Holliday junctions, and biased resolution of these double Holliday junctions into crossovers. The position of the initiating DSB site is indicated by a green vertical line. Left: DNA synthesis of the invading end only, followed by limited branch migration and further processing, results in a crossover with hDNA in each chromatid and on opposite sides of the DSB. If branch migration is more extensive, moving the single-end invasion intermediate away from the initiating DSB site, resolution results in a crossover with hDNA on one chromatid and on the left side of the DSB. Centre: DNA synthesis of the captured end only, followed by branch migration and further processing, results in a crossover with hDNA on one chromatid and on the right side of the DSB. Right: DNA synthesis of both the invading end and the captured end, followed by branch migration and further processing, results in a crossover with hDNA on one chromatid and on both sides of the DSB. Note that branch migration is depicted as unidirectional (to the right) for simplicity but can occur in the opposite direction as well. Branch migration is also possible after annealing.
Which genetic factors are involved?
Are non-Mendelian segregation events (gene conversions) genetically separable from Mendelian ones (crossovers)? The short answer is, not really, apart from one partial exception. Genetic factors which direct or influence the formation and repair of DSBs will affect the frequency of both non-Mendelian and Mendelian segregation events, because these genetic outcomes are determined by the amount and type of meiotic recombination occurring at any given locus. Even the mismatch repair factors MutSα (Msh2-Msh6), Mutsβ (Msh2-Msh3), and MutLα (Mlh1-Pms1), which deal with mismatches in hDNA to produce gene conversions, and modulate the number of non-Mendelian segregation events at polymorphic sites by antirecombination, do also influence overall crossover frequency (Martini et al. 2011; Brown et al. 2019; Ahuja et al. 2021; Cooper et al. 2021). Having said this, the frequency of gene conversion and crossover outcomes can be differentially affected by experimental conditions and certain mutant backgrounds (Rockmill et al. 2013; Brown et al. 2020). This can be explained by changes in DSB frequency and in the way these DSBs are subsequently repaired into crossovers and non-crossovers (Rockmill et al. 2013; Protacio et al. 2022).
The only notable exception of a dedicated factor controlling non-Mendelian segregation is the MutLβ (Mlh1-Mlh2) complex in S. cerevisiae which restricts the tract lengths of gene conversion and PMS in cooperation with the meiosis-specific DNA helicase Mer3, in the context of both crossovers and non-crossovers (Duroc et al. 2017). The authors hypothesise that limiting the length of non-Mendelian segregation events might be necessary to avoid genome integrity issues, due to the high number of DSBs which are simultaneously made and repaired during meiosis (Duroc et al. 2017). They also suggest that too much gene conversion could be detrimental to the genetic diversity within a sexually reproducing population in the long run, as it might destroy favourable allele combinations and, especially in mammals, could cause the extinction of hotspot sequences (Cole et al. 2012; Duroc et al. 2017).
Conclusion
There are many aspects about the generation of non-Mendelian and Mendelian meiotic events that are still enigmatic. The extent to which the mechanism(s) governing non-Mendelian segregation events, gene conversion and PMS, are evolutionarily conserved is also not fully elucidated. There are clear differences between species. For example, gene conversion tracts in S. cerevisiae are substantially longer than in multicellular eukaryotes, and some factors regulating features of non-Mendelian events in S. cerevisiae are not conserved in Sz. pombe (Mlh2, Mer3). The large evolutionary distance between the two model yeasts and their usefulness as experimental systems in meiosis research could be exploited comparatively to shed light on the conservation of the mechanisms enabling non-Mendelian segregation. Clearly, differences between homologous chromosomes in individuals within natural populations, influence where gene conversion events occur during meiosis, and whether they can be detected. This then also affects whether and how they contribute to genetic diversity in a given population. It will be important to bridge the knowledge gaps between molecular genetics, population genetics, and evolutionary biology, to arrive at a truly integrated model of meiotic recombination and its long-term role in generating genetic diversity.
References
Ahuja JS, Harvey CS, Wheeler DL, Lichten M (2021) Repeated strand invasion and extensive branch migration are hallmarks of meiotic recombination. Mol Cell 81:4258–4270.e4
Alani E, Reenan RA, Kolodner RD (1994) Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics 137:19–39
Allers T, Lichten M (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106:47–57
Almanzar DE, Gordon SG, Rog O (2021) Meiotic sister chromatid exchanges are rare in C. elegans. Curr Biol 31:1499–1507.e3
Bateson W (1909) Mendel’s principles of heredity. Cambridge University Press, Cambridge
Baur M, Hartsuiker E, Lehmann E, Ludin K, Munz P, Kohli J (2005) The meiotic recombination hot spot ura4A in Schizosaccharomyces pombe. Genetics 169:551–561
Brown SD, Audoynaud C, Lorenz A (2020) Intragenic meiotic recombination in Schizosaccharomyces pombe is sensitive to environmental temperature changes. Chromosome Res 28:195–207
Brown SD, Mpaulo SJ, Asogwa MN, Jézéquel M, Whitby MC, Lorenz A (2019) DNA sequence differences are determinants of meiotic recombination outcome. Sci Rep 9:16446
Brunswik H (1926) Die Reduktionsteilung bei den Basidiomyzeten. Z für Botanik 18:481–498
Case ME, Giles NH (1958) Evidence from tetrad analysis for both normal and aberrant recombination between allelic mutants in Neurospora crassa. Proc Natl Acad Sci USA 44:378–390
Cejka P, Symington LS (2021) DNA end resection: mechanism and control. Annu Rev Genet 55:285–307
Chen SY, Tsubouchi T, Rockmill B, Sandler JS, Richards DR, Vader G et al. (2008) Global analysis of the meiotic crossover landscape. Dev Cell 15:401–415
Cole F, Baudat F, Grey C, Keeney S, de Massy B, Jasin M (2014) Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat Genet 46:1072–1080
Cole F, Keeney S, Jasin M (2010) Comprehensive, fine-scale dissection of homologous recombination outcomes at a hot spot in mouse meiosis. Mol Cell 39:700–710
Cole F, Keeney S, Jasin M (2012) Preaching about the converted: how meiotic gene conversion influences genomic diversity. Ann NY Acad Sci 1267:95–102
Comeron JM, Ratnappan R, Bailin S (2012) The many landscapes of recombination in Drosophila melanogaster. PLoS Genet 8:e1002905
Cooper TJ, Crawford MR, Hunt LJ, Marsolier-Kergoat M-C, Llorente B, Neale MJ (2021). Mismatch repair impedes meiotic crossover interference. bioRxiv https://doi.org/10.1101/480418.
Cromie GA, Rubio CA, Hyppa RW, Smith GR (2005) A natural meiotic DNA break site in Schizosaccharomyces pombe is a hotspot of gene conversion, highly associated with crossing over. Genetics 169:595–605
Drouaud J, Khademian H, Giraut L, Zanni V, Bellalou S, Henderson IR et al. (2013) Contrasted patterns of crossover and non-crossover at Arabidopsis thaliana meiotic recombination hotspots. PLoS Genet 9:e1003922
Duroc Y, Kumar R, Ranjha L, Adam C, Guérois R, Md Muntaz K et al. (2017) Concerted action of the MutLβ heterodimer and Mer3 helicase regulates the global extent of meiotic gene conversion. eLife 6:e21900
Fogel S, Hurst DD (1967) Meiotic gene conversion in yeast tetrads and the theory of recombination. Genetics 57:455–481
Fogel S, Mortimer R, Lusnak K, Tavares F (1979) Meiotic gene conversion: a signal of the basic recombination event in yeast. Cold Spring Harb Symp Quant Biol 43:1325–1341
Gergelits V, Parvanov E, Simecek P, Forejt J (2021) Chromosome-wide characterization of meiotic noncrossovers (gene conversions) in mouse hybrids. Genetics 217:iyaa013
Gutz H (1971) Site specific induction of gene conversion in Schizosaccharomyces pombe. Genetics 69:317–337
Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB (2001) Molecular evidence for the early colonization of land by fungi and plants. Science 293:1129–1133
Hunter N (2015) Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol 7:a016618
Hunter N, Borts RH (1997) Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev 11:1573–1582
Jeffreys AJ, May CA (2004) Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat Genet 36:151–156
Johnson D, Crawford M, Cooper T, Claeys Bouuaert C, Keeney S, Llorente B et al. (2021) Concerted cutting by Spo11 illuminates meiotic DNA break mechanics. Nature 594:572–576
Juang RH, Mccue KF, Ow DW (1993) Two purine biosynthetic enzymes that are required for cadmium tolerance in Schizosaccharomyces pombe utilize cysteine sulfinate in vitro. Arch Biochem Biophys 304:392–401
Kan F, Davidson MK, Wahls WP (2011) Meiotic recombination protein Rec12: functional conservation, crossover homeostasis and early crossover/non-crossover decision. Nucleic Acids Res 39:1460–1472
Kon N, Krawchuk MD, Warren BG, Smith GR, Wahls WP (1997) Transcription factor Mts1/Mts2 (Atf1/Pcr1, Gad7/Pcr1) activates the M26 meiotic recombination hotspot in Schizosaccharomyces pombe. Proc Natl Acad Sci USA 94:13765–13770
Lam I, Keeney S (2015) Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol 7:a016634
Lao JP, Oh SD, Shinohara M, Shinohara A, Hunter N (2008) Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell 29:517–524
Leupold U (1958) Studies on recombination in Schizosaccharomyces pombe. Cold Spring Harb Symp Quant Biol 23:161–170
Lichten M, Goldman AS (1995) Meiotic recombination hotspots. Annu Rev Genet 29:423–444
Lindegren CC (1949) Chromosome maps of Saccharomyces. Hereditas 35:338–355
Lindegren CC (1958) Priority in gene-conversion. Experientia 14:444–445
Lorenz A (2017) Modulation of meiotic homologous recombination by DNA helicases. Yeast 34:195–203
Lorenz A, West SC, Whitby MC (2010) The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic Acids Res 38:1866–1873
Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454:479–485
Mancera E, Bourgon R, Huber W, Steinmetz LM (2011) Genome-wide survey of post-meiotic segregation during yeast recombination. Genome Biol 12:R36
Marsolier-Kergoat MC, Khan MM, Schott J, Zhu X, Llorente B (2018) Mechanistic view and genetic control of DNA recombination during meiosis. Mol Cell 70:9–20
Martini E, Borde V, Legendre M, Audic S, Regnault B, Soubigou G et al. (2011) Genome-wide analysis of heteroduplex DNA in mismatch repair-deficient yeast cells reveals novel properties of meiotic recombination pathways. PLoS Genet 7:e1002305
Miller DE, Takeo S, Nandanan K, Paulson A, Gogol MM, Noll AC et al. (2012) A whole-chromosome analysis of meiotic recombination in Drosophila melanogaster. G3 Genes, Genomes, Genet 2:249–260
Morgan TH (1910) Chromosomes and heredity. Am Nat 44:449–496
Oke A, Anderson CM, Yam P, Fung JC (2014) Controlling meiotic recombinational repair - specifying the roles of ZMMs, Sgs1 and Mus81/Mms4 in crossover formation. PLoS Genet 10:e1004690
Osman F, Dixon J, Doe CL, Whitby MC (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol Cell 12:761–774
Petes TD (2001) Meiotic recombination hot spots and cold spots. Nat Rev Genet 2:360–369
Ponticelli AS, Sena EP, Smith GR (1988) Genetic and physical analysis of the M26 recombination hotspot of Schizosaccharomyces pombe. Genetics 119:491–497
Prieler S, Chen D, Huang L, Mayrhofer E, Zsótér S, Vesely M et al. (2021) Spo11 generates gaps through concerted cuts at sites of topological stress. Nature 594:577–582
Protacio RU, Mukiza TO, Davidson MK, Wahls WP (2022) Molecular mechanisms for environmentally induced and evolutionarily rapid redistribution (plasticity) of meiotic recombination. Genetics 220:iyab212
Radford SJ, Sabourin MM, McMahan S, Sekelsky J (2007) Meiotic recombination in Drosophila Msh6 mutants yields discontinuous gene conversion tracts. Genetics 176:53–62
Rébora K, Desmoucelles C, Borne F, Pinson B, Daignan-Fornier B (2001) Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol Cell Biol 21:7901–7912
Reiter LT, Hastings PJ, Nelis E, De Jonghe P, Van Broeckhoven C, Lupski JR (1998) Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet 62:1023–1033
Resnick MA (1976) The repair of double-strand breaks in DNA: a model involving recombination. J Theor Biol 59:97–106
Rieger R, Michaelis A, Green M (1991) Glossary of genetics - classical and molecular, 5th edn. Springer-Verlag Berlin Heidelberg New York, Berlin, Heidelberg
Robert VJ, Davis MW, Jorgensen EM, Bessereau J-L (2008) Gene conversion and end-joining-repair double-strand breaks in the Caenorhabditis elegans germline. Genetics 180:673–679
Rockmill B, Lefrançois P, Voelkel-Meiman K, Oke A, Roeder GS, Fung JC (2013) High throughput sequencing reveals alterations in the recombination signatures with diminishing Spo11 activity. PLoS Genet 9:e1003932
Schär P, Baur M, Schneider C, Kohli J (1997) Mismatch repair in Schizosaccharomyces pombe requires the mutL homologous gene pms1: molecular cloning and functional analysis. Genetics 146:1275–1286
Schär P, Kohli J (1993) Marker effects of G to C transversions on intragenic recombination and mismatch repair in Schizosaccharomyces pombe. Genetics 133:825–835
Schär P, Munz P, Kohli J (1993) Meiotic mismatch repair quantified on the basis of segregation patterns in Schizosaccharomyces pombe. Genetics 133:815–824
Schuchert P, Kohli J (1988) The ade6-M26 mutation of Schizosaccharomyces pombe increases the frequency of crossing over. Genetics 119:507–515
Spies M, Fishel R (2015) Mismatch repair during homologous and homeologous recombination. Cold Spring Harb Perspect Biol 7:a022657
Steiner WW, Schreckhise RW, Smith GR (2002) Meiotic DNA breaks at the S. pombe recombination hot spot M26. Mol Cell 9:847–855
Steiner WW, Smith GR (2005a) Natural meiotic recombination hot spots in the Schizosaccharomyces pombe genome successfully predicted from the simple sequence motif M26. Mol Cell Biol 25:9054–9062
Steiner WW, Smith GR (2005b) Optimizing the nucleotide sequence of a meiotic recombination hotspot in Schizosaccharomyces pombe. Genetics 169:1973–1983
Sun Y, Ambrose JH, Haughey BS, Webster TD, Pierrie SN, Muñoz DF et al. (2012) Deep genome-wide measurement of meiotic gene conversion using tetrad analysis in Arabidopsis thaliana. PLoS Genet 8:e1002968
Surtees JA, Argueso JL, Alani E (2004) Mismatch repair proteins: Key regulators of genetic recombination. Cytogenet Genome Res 107:146–159
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (1983) The double-strand-break repair model for recombination. Cell 33:25–35
Toraason E, Horacek A, Clark C, Glover ML, Adler VL, Premkumar T et al. (2021) Meiotic DNA break repair can utilize homolog-independent chromatid templates in C. elegans. Curr Biol 31:1508–1514.e5
Wahls WP, Davidson MK (2012) New paradigms for conserved, multifactorial, cis-acting regulation of meiotic recombination. Nucleic Acids Res 40:9983–9989
von Wettstein F (1924) Morphologie und Physiologie des Formwechsels der Moose auf genetischer Grundlage. I. Z für Indukt Abstamm- und Vererbungslehre 33:1–236
White MA, Petes TD (1994) Analysis of meiotic recombination events near a recombination hotspot in the yeast Saccharomyces cerevisiae. Curr Genet 26:21–30
Whitehouse HLK, Hastings PJ (1965) The analysis of genetic recombination on the polaron hybrid DNA model. Genet Res 6:27–92
Wijnker E, Velikkakam James G, Ding J, Becker F, Klasen JR, Rawat V et al. (2013) The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife 2:e01426
Winkler H (1930) Die Konversion der Gene: eine vererbungstheoretische Untersuchung. Gustav Fischer, Jena
Zahn-Zabal M, Kohli J (1996) The distance-dependence of the fission yeast ade6-M26 marker effect in two-factor crosses. Curr Genet 29:530–536
Zickler D, Kleckner N (2015) Recombination, pairing, and synapsis of homologs during meiosis. Cold Spring Harb Perspect Biol 7:a016626
Zierhut C, Diffley JFX (2008) Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27:1875–85
Acknowledgements
We are grateful to Pete Carlton and Diana Libuda for discussing meiotic recombination in C. elegans. We acknowledge funding by the EASTBIO doctoral training partnership of the Biotechnology and Biological Sciences Research Council UK (BBSRC) [grant No. BB/M010996/1].
Author information
Authors and Affiliations
Contributions
AL conceived the article, and was responsible for conducting literature research, drafting the main text, and revising the figures and figure legends. SJM was responsible for conducting literature research, drafting the figures and figure legends, and revising the main text. Both authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lorenz, A., Mpaulo, S.J. Gene conversion: a non-Mendelian process integral to meiotic recombination. Heredity 129, 56–63 (2022). https://doi.org/10.1038/s41437-022-00523-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00523-3
