
Abstract
Clupeoid fish can be considered excellent candidates to understand the role of mitochondrial DNA in adaptive evolution, as they have colonized different habitats (marine, brackish, freshwater, tropical and temperate regions) over millions of years. Here, we investigate patterns of tRNA location, codon usage bias, and lineage-specific diversifying selection signals to provide novel insights into how evolutionary improvements of mitochondrial metabolic efficiency have allowed clupeids to adapt to different habitats. Based on whole mitogenome data of 70 Clupeoids with a global distribution we find that purifying selection was the dominant force acting and that the mutational deamination pressure in mtDNA was stronger than the codon/amino acid constraints. The codon usage pattern appears evolved to achieve high translational efficiency (codon/amino acid-related constraints), as indicated by the complementarity of most codons to the GT-saturated tRNA anticodon sites (retained by deamination-induced pressure) and usage of the codons of the tRNA genes situated near to the control region (fixed by deamination pressure) where transcription efficiency was high. The observed shift in codon preference patterns between marine and euryhaline/freshwater Clupeoids indicates possible selection for improved translational efficiency in mitochondrial genes while adapting to low-salinity habitats. This mitogenomic plasticity and enhanced efficiency of the metabolic machinery may have contributed to the evolutionary success and abundance of Clupeoid fish.
Similar content being viewed by others

Introduction
The order Clupeiformes includes sardines, herrings, anchovies, and other relatives in the two major suborders Denticipitoidei and Clupeoidei. More than 390 species belonging to five families, mainly Clupeidae, Engraulidae, Chirocentridae, Pristigasteridae and Sundasalangidae have been reported (Lavoue et al. 2014). Clupeiformes exhibit exemplary diversity and trophic diversification (Egan et al. 2018) and inhabit a wide variety of habitats such as the open ocean, coastal areas, estuaries, freshwater rivers and lakes in tropical and temperate regions of all continents except Antarctica. The greatest diversity in clupeiform fish is found in the Indo-West Pacific region with a high level of endemism (Lavoue et al. 2014). A mitogenomic phylogeographic study of Clupeoids suggested that the East Tethys sea region is the Indo-West Pacific progenitor region where the initial diversification of Clupeoids occurred during the Cretaceous/Paleogene (Lavoue et al. 2013). Subsequently, multiple independent marine/freshwater/tropical/temperate transitions accelerated the evolutionary diversification of Clupeoids in the world’s oceans (Ganias 2014).
The mitochondrial genome, or mtDNA, is the genetic material of the mitochondrion, the organelle that functions as the powerhouse of eukaryotic cells. A typical animal mtDNA encodes 13 proteins, 2 ribosomal RNA (rRNA) genes and 22 tRNA genes in its heavy (H) and light (L) strands (Boore 1999). A gene arrangement pattern has been conserved in vertebrate mtDNA with some exceptions. The metabolic performance of organisms is affected by mutations in mitochondrial DNA (mtDNA) (Lajbner et al. 2018) and therefore purifying selection is an important driving force for their evolution (Jacobsen et al. 2016). Despite this, there is evidence of directional or episodic positive selection in response to shifts in selection pressures such as hypoxia (Da Fonseca et al. 2008), heat stress (Morales et al. 2015), cold stress (Stier et al. 2014), and nutrient availability (Da Fonseca et al. 2008) in several organisms (Garvin et al. 2015a, 2015b; Lajbner et al. 2018; Teske et al. 2019). The evolutionary dynamics of the mitogenome are also influenced by indirect selection by the nuclear genome due to mitonuclear co-evolution (Morales et al. 2016). Maintaining optimal mitonuclear association in the OXPHOS system is critical as mismatches have adverse effects such as reduced lifespan, fecundity, reduced metabolic rate and disease (Dowling et al. 2008; Gershoni et al. 2014; Mossman et al. 2019). Adaptive evolution of mtDNA in response to habitat changes has been reported in humans (Mishmar et al. 2003; Ruiz-Pesini et al. 2004; Balloux et al. 2009), Drosophila (Ballard et al. 2007; Camus et al. 2017), Atlantic cod, Atlantic salmon, Pacific salmon and Killer whale populations (Foote et al. 2011; Garvin et al. 2011; Consuegra et al. 2015). Evidence for adaptive evolution in the mtDNA suggests its possible role in the radiation, successful diversification and adaptation of fish to different habitats such as marine, euryhaline, cold and warm waters (Garvin et al. 2015a, b; Morales et al. 2016; Carapelli et al. 2019).
The mitochondrial H strand is first replicated from the H strand origin of replication (Ori OH) within the control region according to the mitochondrial DNA replication model. The original H-strand is then exposed as a single strand, which acts as a lagging strand during the synthesis of the L-strand. The L strand is replicated from the L strand origin of replication (Ori OL), complementary to the original H strand (Clayton 1991). The DNA sequences exposed as single-stranded for a long time (during replication and transcription) are prone to spontaneous deamination mutations (which are more common on single-stranded than on double-stranded DNA) mainly in the regions distant from the OL towards L-strand replication. Deamination often occurs in single-stranded DNA exposed during replication or transcription, resulting in a C-to-T mutation on the H-strand and a consequent G-to-A mutation on the L-strand (Shadeland Clayton 1997; Lowell and Spiegelman 2000). The regions close to the control region are characterized by a high rate of expression and deamination mutations, which is attributed to the presence of transcription initiation and Ori-OH sites, respectively (Xia 2005; Satoh et al. 2010). The high structural conservation in vertebrate mtDNA has been proposed as a result of some selection constraints such as mutational pressure and translational selection (Xia 2005; Satoh et al. 2010). Therefore, tRNA anticodon sites, tRNA gene order, codon usage, and base pair composition in fish mitogenomes may be under constant mutational pressure and translational selection (Xia 2005; Satoh et al. 2010).
Codon usage bias (synonymous codons not used with equal frequency) plays many important roles in RNA processing, protein translation and protein folding (Pernaand Kocher 1995; McLean et al. 1998). Two main hypotheses explain codon usage bias; The selection hypothesis is based on the concept that codon usage determines the efficiency and/or fidelity of protein expression (Xia 2005; Satoh et al. 2010). Thus, codon bias is created and maintained by natural selection. In contrast, the mutational or neutral hypothesis proposes that the codon bias is due to the non-random mutational patterns (Xia 2005; Satoh et al. 2010). The selective and neutral codon usage hypotheses contradict each other, but both mechanisms play a role in codon usage patterns within and between genomes. Comparative vertebrate mitogenomics suggested that codon usage bias is maintained by strand-specific mutational bias and biased codon usage drives the evolution of tRNA anticodons (Xia 2005). The transcription rate is high near the mtDNA control region, and it has been suggested that the tRNA genes corresponding to commonly used codons are closer to the control region for efficient transcription (Satoh et al. 2010). Besides, the tRNA loci exposed as single-stranded for a longer time have more guanine and thymine in their anticodon sites (Satoh et al. 2010). However, the dynamics of the evolutionary pattern of tRNA anticodon sites, tRNA gene order, codon usage, and base-pair composition of the vertebrate mitogenome is still ambiguous.
The two main epithelial cells in fish gills, pavement cells (PVCs) and mitochondrial rich cells (MRCs), play key roles in ionic and water balance (homeostasis) in fish migrating between freshwater (FW) and seawater (SW) (Evans et al. 2005; Lai et al. 2015). Adaptation to such habitat changes was achieved through an increase in the number of mitochondria in the cells (Schreiber and Specker 2000) together with an increase in mitochondrial coupling (Brijs et al. 2017) and expression (Xia 2005). Radical amino acid changes or specific substitutions in mtDNA would have enhanced mitochondrial coupling and conferred an advantage in habitat diversification (Foote et al. 2011; Garvin et al. 2011; Consuegra et al. 2015; Sebastian et al. 2020). Signals of positive selection in the mitogenome associated with major changes in physiology or ecology, such as the origins of electrogenesis in fish and the evolution of powered flight in bats have been reported by using comparative genomic analysis (Shen et al. 2010; Elbassiouny et al. 2020). The diversity of habitats colonized by Clupeoid fish, together with a high degree of endemism and a large population size (less effect of genetic drift) make them excellent candidates for studies of adaptive evolution and diversifying selection on the mitogenome.
In the present study, we analyzed the signals of selective forces on the mtDNA coding regions of fish of the suborder Clupeoidei to understand the selective pressures on the mitogenome associated with habitat changes and the resulting higher energy demands. The whole mitogenomes of 70 Clupeoids were analyzed for the pattern of codon usage and nucleotide substitution between lineages to reveal events of positive selection and the patterns of codon evolution. The results of these studies provided important insights into the dynamics of mitochondrial genome evolution during the diversification of Clupeoid fish from their marine ancestor in the Indo-West Pacific Ancestor Region to different world ocean habitats such as marine, euryhaline, fresh, cold, and warm water.
Materials and methods
The complete mitochondrial genomes of 70 Clupeoids (from the families available in NCBI, GenBank) were selected for analysis (Lavoue et al. 2013) (Supplementary file_1_Table S4). The spatial distribution data of all selected species were obtained from Lavoue et al. 2014 and FishBase (www.fishbase.in). The mitogenome sequence of Denticeps clupeoids (the sister group of the Clupeoids) was chosen as the outgroup. Protein-coding gene regions were aligned in MEGA7 (Kumar et al. 2016) with CLUSTALW and a concatenated dataset was created. A maximum likelihood phylogenetic tree was then constructed using the General Time Reversible (GTR + I) model of substitution selected using the J-model test (Posada 2008) with 1000 bootstrap replication and four gamma categories. Subsequent analyzes were performed on this tree.
Codon and amino acid usage was determined for each protein-encoding gene after excluding the partial or full stop codons at its end in MEGA7 and Geneious R7 (Kearse et al. 2012). The mean of the GC content in the first (GC1) and second (GC2) positions of the codons (GC12) was used for the analysis of the neutrality plot (GC12 vs. GC3) (Sueoka 1988). The nucleotide bias or skew was calculated as (A-T)/(A + T) or (G-C)/(G + C). The effective number of codons (ENc) was estimated using DAMBE 5 (Xia 2013) and used as a measure of codon usage bias in genes (Wright 1990). The relative synonymous codon usage (RSCU) was calculated in MEGA7. To avoid pseudo-replication, closely related species in each lineage were grouped according to their habitat characteristics (freshwater/euryhaline and marine) (each group can be viewed as an evolutionarily independent entity with the same habitat characteristics) and average nucleotide composition (A, T, G and C of the three codon position) and RSCU (of the 60 codons) of each unit were used for statistical analysis. The Kolmogorov-Smirnov test was used to analyze the normality of continuous variables (nucleotide composition and RSCU). Point-biserial correlation and cross-tabulation analyzes (and chi-square statistics) were performed to test the correlation/association between nucleotide composition and RSCU in the protein-coding genes (concatenated data) with their habitat (freshwater/euryhaline and marine). The nucleotide composition and RSCU of the protein-coding genes (concatenated data) of each Clupeoid species were considered as continuous variables and their habitat characteristics (freshwater/euryhaline and marine) as dichotomous variables. The coefficient (r) and p value were calculated for each test using point biserial correlation analysis in the R statistics package (R Core Team 2021). We also performed crosstab and chi-square statistical analysis because some of the continuous variable data deviated slightly from the normal distribution. A cross-tabulation was made by dividing the percentage of nucleotide composition and RSCU into four categories: low, medium-low, medium-high and high. The contingency table was then visualized using gplots (function balloon plot) in the R statistics package. X-square (χ2) and p value were calculated for each test using the chi-square test (function chisq.test) in the R-statistics package.
Positive selection for the 13 protein-coding genes of Clupeoids was analyzed using three codon-based selection analysis algorithms; Fast Unconstrained Bayesian Approximation (FUBAR), Mixed Effects Model of Evolution (MEME) and TreeSAAP. Both MEME and FUBAR are site-based detection methods (available in DATA MONKEY) (Pond and Frost 2005), allow synonymous rate variation from site to site, and use likelihood ratio tests (LRTs) at individual sites to assess the significance of positive selection. The MEME model analyzes the distribution of synonymous and non-synonymous substitution rates from site to site and from branch to branch at a site (episodic selection) (Murrell et al. 2012). While FUBAR uses a Bayesian approach to derive non-synonymous (dN) and synonymous (dS) substitution rates per site for a given coding alignment and corresponding phylogeny (pervasive selection) (Murrell et al. 2013). For each method we have chosen a threshold p value; p < 0.05 for MEME and posterior probability >0.9 for FUBAR. TreeSAAP (Woolley et al. 2003) was used to identify selected sites and changes in the physicochemical properties of amino acids caused by substitutions at selected sites. The amino acid sites identified as candidate sites for positive selection with FUBAR, MEME and TreeSAAP (i.e., the sites commonly identified in the three methods) and those associated with the internal branches were used as candidate sites for subsequent analysis since the sites associated with terminal branches may underlie relaxed purifying selection or the fixation of mildly deleterious changes through genetic drift (Jacobsen et al. 2016).
The number of radical amino acid changes and synonymous substitutions associated with each branch of the Clupeoid mitogenomic phylogenetic tree was extracted from TreeSAAP analyses. A Pearson correlation analysis was performed to identify the role of two potential evolutionary forces: positive selection and neutral/slightly deleterious changes behind the observed radical physiochemical amino acid changes associated with each branch. The average of the number of radical amino acid changes and synonymous changes of each evolutionarily independent entity was used for correlation analysis to avoid pseudo-replication. Both datasets followed the assumptions of normal distribution, linearity, and homoscedasticity. We tested the hypothesis that there is no significant correlation between the number of radical amino acid changes and synonymous changes (i.e., the number of radical physiochemical amino acid changes does not reflect the evolutionary distance generated by neutral/slightly deleterious changes).
3D homology models of the protein subunits showing candidate sites of positive selection were generated by the SWISS MODEL server (Schwede et al. 2003) using appropriate subunits of the protein structure with Bos taurus as a template. The candidate sites were then mapped onto the three-dimensional structure.
Results
The geographical distribution of the Clupeoids of the present study is shown in Fig. 1. The maximum likelihood mitogenomic tree showed six moderately to strongly supported monophyletic groups (bootstrap value range of 65–100) within the order Clupeiformes (Fig. 2, Supplementary file_2_Fig. S1). The family Clupeidae and its five subfamilies were not monophyletic. Only three of the nine families currently recognized (by multiple morphological characters): Engraulidae, Pristigasteridae, and Dussumieriidae formed well-supported monophyletic groups. Four other lineages belonged to mixed taxa (designated lineages 1 to 4), similar to previous studies (Lavoue et al. 2013; Lavoue et al. 2014). They represented lineages formed by the major second and third dispersal events in Clupeoids, respectively (Lavoue et al. 2013). The relationship between other lineages was moderately to strongly supported (bootstrap value range of 65–100), resulting in the same major lineages observed in previous studies (Lavoue et al. 2013; Lavoue et al. 2014). The moderate bootstrap support may be the result of a weak phylogenetic signal in the mtDNA and incomplete sampling of Clupeoid taxa (the whole mitogenome of many Clupeoid taxa is not available). To avoid the influence of tree topology uncertainties on the selection analysis, we restricted our analysis and interpretation to the main lineages (lineages associated with basal nodes; nodes 71–78) that have sufficient statistical support from fossil distributions, phylogenetic inferences, and biogeographical reconstructions (Lavoue et al. 2013, 2014).

(A) 70 Clupeoids used for the study (AU South Australia, EA East Atlantic, EP East Pacific, IWP Indo-West Pacific, NA North west Atlantic, NE North East Atlantic, NP North Pacific, NZ New Zealand, PC Ponto Caspian, SA South Africa, SS South South America, WA West Atlantic) and (B) their biogeographical distribution. The spatial distribution data of the species were taken from the FishBase database (www.fishbase.in) and Lavoue et al. 2014.

Denticeps clupeoides was used as the outgroup. Bootstrap values are indicated in the bold and phylogenetic distance in gray color. Shapes in tips of phylogeny indicate salinity preference: marine (black circle), euryhaline (white square), and freshwater (white circle). Temperate water species are indicated with “Tem”. The black arrow indicates the transition from marine to euryhaline or freshwater environments.
Nucleotide composition, codon usage, tRNA anticodon composition, and tRNA gene position
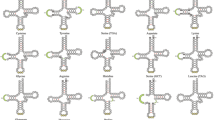
We observed a gradient in the arrangement of genes and amino acid composition relative to the position of the origin of replication (Ori L and Ori H), control region (CR), and codon usage in the mitogenome of Clupeoids (Fig. 3b, c). A significant correlation was obtained between the GT content in the H-strand tRNA anticodon sites and the estimated duration of single-strand exposure/position along the direction of H-strand replication (Supplementary file_2_Fig. S4). Similarly, a moderate correlation is found between the GT content in the L-strand tRNA anticodon sites and its position between OH-OL and OL-OH along the H-direction, with the exception of tRNA Pro (Supplementary file_2_Fig. S4). The tRNAs with anticodons of commonly used codons have been positioned near the control region where the transcription efficiency is high (Satoh et al. 2010), and among these, the tRNA with anticodons corresponding to the hydrophobic amino acids is common (Fig. 3c, Supplementary file_2_Fig. S3). The wobble nucleotide position of the tRNA anticodons in the Clupeoids mtDNA showed a strong G and U bias and a very strong anti-G bias in the 3rd codon position of all synonymous codon families except arginine (CGG) and methionine (AUG) (Fig. 3c, Supplementary file_3_Fig. S11).

A Percentage of A, T, G, and C in mitogenome, protein-coding genes, merged protein-coding gene, 12 S rRNA, 16 S rRNA, and 22 tRNAs. B Schematic diagram of the mtDNA replication based on the displacement model of replication. Mitogenome map of a vertebrate with protein-coding genes, tRNAs, rRNAs, and D-loop regions in different colors are represented in inner circles. The direction of H and L-strand replication from OH and OL based on the displacement model is indicated in outer circles. C tRNA genes, its codon, anticodon, and order of distribution on H and L strand of Clupeoid mtDNA.
The base composition of both the L- (rich in A + C) and H-strand (rich in G + T) genes is consistent with the strand-specific mutational bias observed in the vertebrate mitogenome (Boore 1999). The strand-specific base composition was also observed in tRNA (Supplementary file_2_Fig. S2). The distribution of A and G coding genes in the marine lineages compared to other lineages (freshwater and brackish water) showed a remarkable difference. All marine species showed a shift to high G (18–29%) and low A (20–25%) compared to euryhaline and freshwater fish (A 26–29% and G 14–17%) (Fig. 4). Even though there is no notable difference in the distribution of nucleotides at the 1st and 2nd codon positions between species, the 3rd codon position showed a clear divergence, particularly in the composition of adenine (A3) and guanine (G3). Both freshwater and euryhaline species preferred A over G, while the marine lineages preferred G over A in the third codon position, except in Engraulidae (Fig. 4).

Species in Engraulidae and all the other species are sorted separately according to their habitat and salinity preference indicated as Marine, Euryhaline, and Freshwater. Color scale indicates A, T, G, and C content at different codon positions of merged protein-coding genes for each species.
High/very high associations in cross-tabulation analysis (n = 39, p < 0.001) and a significant negative correlation in point biserial correlation (n = 39, p < 0.01) were found between the distribution of nucleotide G on the 3rd codon position of the mitogenomic protein-coding genes and their habitat (i.e., freshwater/euryhaline) except in Engraulidae. In contrast, a high/very high association and positive correlation (n = 39, p < 0.001; n = 39, p < 0.03) with marine species was observed for A at the 3rd codon position (Table 1), supporting the preference of A3 over G3 in freshwater and euryhaline species and G3 over A3 in marine lineages (Table 1). Although not high, a significant association (A3: n = 12, p < 0.05, G3: n = 12, p < 0.05) and correlation was also found in G3 and A3 of Clupeoids in Engraulidae (A3: n = 12, p < 0.01, G3: n = 12, p < 0.001) (Table 1). The trend of nucleotide usage can be observed in the balloon plot (Supplementary file_3_Fig. S15).
The Clupeoid mitogenome is rich in codons encoding leucine (Leu ~16%), followed by alanine (Ala ~9%) and threonine (Thr 8.5%). Asparagine, arginine, lysine (2%) and cysteine (~0.8%) were the least frequent. Although the overall amino acid composition of the concatenated gene data set showed no differences between species, wobble nucleotide usage of the tRNA anticodon varied between species. Relative Synonymous Codon Usage (RSCU) analysis revealed that the Clupeoid L-strand -encoded genes preferred codons with nucleotide A and C over G and T at their 3rd codon position, while the H-strand encoded ND6 preferred T and G over C and A, consistent with the skewing of nucleotide composition in the complete mitogenome (Supplementary file_3_Fig. S11 & file_3). The RSCU distribution values were very low for codons with G at the 3rd position in most freshwater lineages (mean 0.29), followed by euryhaline (mean 0.42) and marine lineages (mean 0.62), while the A at the 3rd position showed the opposite trend. Thus, the observed bias in base composition in the mitogenome is the result of a bias in codon usage. This differential codon usage was not restricted to specific protein-coding genes, as it occurred in most of them. Comparisons of usage of tRNA anticodon and synonymous codon families showed that most codons with the highest RSCU matched the 22 identified tRNAs in the mitogenome (with the exception of tRNA Arg -CCG and tRNA Met -AUG). Most freshwater and euryhaline lineages showed exceptionally high RSCU values for codons fully paired with tRNA anticodon in the mitogenome, with the exception of a few species in Engraulidae, which did not follow this pattern strongly. In general, freshwater, followed by euryhaline species showed a very strong anti-G bias at their 3rd codon position and a strong preference for codon matching with tRNA anticodon (especially codon with A at 3rd position) in mitogenome. In contrast, the marine lineages showed reduced restrictions in anti-G propensity and preference for a codon matching the anti-codon (A at the 3rd position) in the mitogenome. This signals the role of a directed mutation in the observed codon usage pattern of the Clupeoids mitogenome. A significant positive and negative point biserial correlation was obtained for the correlation analysis of the RSCU of the codon with A or G at the 3rd position with the habitat (freshwater/euryhaline and marine) of Clupeoid fish, respectively (Table 1). Both point-biserial correlation and cross-tab testing statistically supported the preference for A3 over G3 in freshwater/euryhaline species and G3 over A3 in marine lineages (A3: n = 39, p < 0.01–0.0001, G3: n = 39, p < 0.01–0.0001 & A3: n = 39, p < 0.01–0.0001, G3: n = 39, p < 0.01–0.0001). Although the association/correlation was less strong, statistically significant support was obtained for all analyzes in the Engraulids as well (A3: n = 12, p < 0.05–0.0001, G3: n = 12, p < 0.05–0.0001 & A3: n = 12, p < 0.05, G3: n = 12, p < 0.05) (Table 1), which was also visible in the balloon plot of the data (Supplementary file_3_Fig. S16).
Neutrality plot analysis (GC12 vs. GC3) (r-value is 0.69) showed that GC12 and GC3 followed a mutational bias model with a moderate correlation between GC12 and GC3. Similar to the RSCU results, the effective number of codons (ENc) ranged from 46.4 to 58.1 (which is lower in freshwater lineages, with the exception of Engraulids and Tenualosa), indicating high codon usage bias in the Clupeoid mitogenomes (Supplementary file_1_Table S1). The standard curve in the ENc plot represents the functional relationship between ENc and GC3 under mutational and selection pressure. When codon usage bias is based entirely on mutation bias (GC3 content), all points lie on the standard curve. All values were above the ENc plot curve (not on the ENc plot curve) in the ENc plot with concatenated gene data set of freshwater/brackish water and seawater fish (Supplementary file_3_Fig. S13). Thus, though mutational bias is the main force shaping the observed codon bias (Chen et al. 2014), other factors such as natural selection, selection by gene length, and expression levels also likely modulate the selection constraints for codon usage bias in the mitogenome (Chen et al. 2014). The Codon Adaptation Index (CIA) value (based on Rattus norvegicus) of Clupeoid mitochondrial protein-encoding genes ranged from 0.5 to 0.6, indicating a comparatively high expression level.
Positive selection
The MEME and FUBAR analyzes showed that the positively selected sites were located in complex 1 (ND1, ND2, ND3, ND4, ND4L, ND5 and ND6), complex 2 (CYTB), complex 4 (CO1, CO2 and CO3) and complex 5 (ATP 6) (Supplementary file_1_Table S2, Supplementary file_1) and positive selection signals were less common than purifying selection. The positively selected sites in Complex I, Complex 2, and Complex 5 were constrained to the predicted internal helical loop (coil) region of their respective proteins (Fig. 5, Supplementary File_2_Fig. S5 and Fig. S6). TreeSAAP analysis revealed that several significant physiochemical amino acid changes occurred with changes in amino acid residues of mitochondrial protein-coding sites (Fig. 6, Supplementary file_2_Fig. S7 and file_4_Table S2). Negative selection predominates in both conservative/moderate (category 1, 2, and 3) and radical changes (category 6, 7, 8) (total properties 23674 (1, 2, 3 +) & 27737 (1, 2, 3 −) and 1751 (6,7,8 +) & 1964 (6,7,8 −)). The highest mean number of positive radical amino acid modifications (0.92, 0.70, and 0.058 mean changes per site, respectively) was found by the proteins ND6, ND2, and ND4, and the lowest by CO3, CYTB, ATP8, and CO1 (0.015, 0.015, 0.013, and 0.008 average changes per site) (Supplementary file_3_Fig. S14). Higher positive radical amino acid modifications were observed in the terminal branches/tips (particularly in anadromous and catadromous species) compared to the inner branches (total properties counts 1034 and 755, respectively). (Fig. 6, Supplementary file_2_Fig. S7). The lineage of the converging temperate water Clupeoids (lineage 2 and 4) has a relatively high number of radical amino acid changes (nodes 77 to 100, 101, 102; 75 to 114). Similarly, the lineage converging in the transition from marine to freshwater also showed a high number of amino acid property changes (Fig. 6, Supplementary file_2_Fig. S7). Pearson correlation analysis showed that the number of radical physiochemical amino acid changes in terminal branches correlated moderately with the number of synonymous changes (R2 = 0.58, p = 0.018). But a marginal correlation was observed for the internal branches (R2 = 0.49, p = 0.054).

A Amino acids at candidate site for positive selection in three subunits of Complex IV. B Individual OXPHOS Complex IV (Homodimer) with mitochondrial encoded subunits are represented in different colors (CO1 in orange, CO2 in yellow, CO3 in magenta and gray structures represent nuclear-encoded subunits) and three-dimensional representation of candidate site for selection in individual core subunits.

In the phylogenetic tree, node numbers are indicated in bold and phylogenetic distance in gray color. Black circles, white circles, and squares in the tree indicate marine, brackish and freshwater species respectively. The color key indicates the average number of radical amino acid property changes (positively selected amino acids) in the 13 protein-coding genes of the mitogenome, associated with each lineage. The left and right sides of the figure indicate radical amino acid property changes associated with internal and terminal branches respectively. The unique and total numbers of radical amino acid changes along with the total number of synonymous substitutions are also indicated.
We selected only the sites commonly identified in the three methods (FUBAR, MEME, and TreeSAAP) and those associated with the internal branches as candidate sites for positive selection to avoid false positives. Candidate sites in complex IV were located in the intrahelical loop (CO1 site #133; CO2 site #227, 230), the transmembrane helix (CO1 site #21, 187, 338; CO2 site #44, 221; CO3 site #47) and β-pleated sheet (CO2 site #9) (Fig. 5, Supplementary file_2_Fig. S6). Furthermore, the amino acid residue reported to participate in key functions and to be involved in the interactions between mitochondrial and nuclear subunits does not overlap with sites undergoing radical changes (Tsukihara et al. 1996; Crofts 2004). Freshwater Clupeoids in lineage 3 carried unique amino acid substitutions in ND2 (site #23, 86) and at site #566 of ND5. Similarly, we identified positively selected/radical amino acid changes in ND4 (site #183), ND5 (site #577), and ND6 (site #118) that are specific for temperate water species in lineages 2 and 4. Amino acid substitution C in ND4 (site #183) and A/T/Q in ND5 (site #577) is specific for lineage 3 and D/A in ND6 is specific for lineage 4. Cytochrome c oxidase (complex IV) was notable with a freshwater-specific substitution (cysteine (C) at site #44) in the CO2 of lineages 1 and 3 (Supplementary file_2_Fig. S8), which was the only habitat-specific substitution in all analyses. Similarly, we identified amino acid changes in ND4, ND5, and ND6 that are specific to temperate water species in lineages 2 and 4. Positively selected/radical amino acid changes in ND4 (site#183), ND5 (site#577), and ND6 (site#118) were specific to temperate water species in lineages 2 and 4. Amino acid substitution C in ND4 (site#183) and A/T/Q in ND5 (site#577) is specific to lineage 3 and D/A in ND6 is specific to lineage 4.
Discussion
The evolutionary diversification of Clupeoids was characterized by multiple and independent transitions between marine/freshwater/tropical/temperate regions (Ganias 2014) during the Cretaceous or Palaeogene period (early Cenozoic era) (Lavoue et al. 2013; Lavoue et al. 2014). The mitogenomic phylogeny of the species used in this study also resulted in the same major lineages observed in previous investigations (Lavoue et al. 2013; Lavoue et al. 2014). The high structural and functional conservation of vertebrate mtDNA may be a consequence of selection constraints resulting from mutation pressure, ensurance of translational efficiency (Xia 2005; Satoh et al. 2010) along with functional constraints as it forms the genetic material of a vital organ (Mitochondria) (Boore 1999; Jacobsen et al. 2016). However, the tRNA location, codon usage bias, and lineage-specific diversifying selection signals observed in the mitogenomes of the present study indicated how mitochondrial metabolic efficiency was improved to meet the challenges in the different habitats where Clupeoids fish colonized. The tRNA anticodon in Clupeoids was saturated with guanine (G) or thymine (T), with the exception of tRNA methionine and proline. There is a gradient in the arrangement of tRNA genes in mtDNA relative to the position of the origin of replication (Ori L and Ori H) and the control region (CR). The evolution of the codon usage pattern towards ensuring high translational efficiency (codon/amino acid-related constraints) was evident from the complementarity of most codons to the GT-saturated tRNA anticodon sites (retained by deamination-induced pressure) and the usage of the codons of the tRNA genes situated near the control region (fixed by deamination-induced pressure) where transcription efficiency is high. The observed shift in codon preference patterns between marine and euryhaline/freshwater Clupeoids could be the result of selection for improved translational efficiency in mitochondrial genes while adapting to low-salinity habitats. A strong codon usage bias observed in freshwater vs. marine lineages suggested the responses of Clupeoid fish to osmotic challenges. The third codon position was characterized by a strong anti-G bias in freshwater, followed by euryhaline fish compared to marine fish, and A at the third codon position recorded the opposite trend. The present study demonstrated that codon usage bias, base composition of tRNA anticodon sites, and tRNA gene order were maintained in the Clupeoid mitogenome by the balance between mutational pressure and translational selection. Shifting the codon usage pattern of fresh/brackish water irradiated Clupeoids may be helpful to adapt to the new environment. mtDNA genes may have undergone directed or episodic positive selection in response to shifts in selection pressure during the adaptation of Clupeoids to different habitats. We observed that purifying selection was the dominant force acting on mitochondrial protein-encoding genes. However, we also observed evidence for positive selection at amino acid sites and radical amino acid changes. Radical amino acid changes were highest in ND6, ND2 and ND4 and lowest in CO3, CYTB, ATP8 and CO1. Some of these could be candidate sites for positive selection in Clupeoids with functional importance for adaptation.
Codon usage bias and translational selection
The mutational bias in Clupeoid mtDNA was evident from the skewing of base composition between genes in the protein-coding regions on their H and L strands (on the L strand C > A~T > G), similar to other vertebrates. Although the first codon position showed no variation in base composition, a low G residue and anti-G bias was evident at the second and third codon positions, respectively. The codon ordering in the protein-coding gene of the L-strand of Clupeoid mtDNA confirmed this evidence, since A and C were preferred at the 3rd codon position with a strong anti-G bias, while the gene on the H-strand showed opposite patterns. The dominant role of mutation bias in the observed codon usage bias was also revealed by the ENc plot (Chen et al. 2014). However, other factors such as natural selection, selection for gene length, and expression levels also likely modulate the selection constraints for codon usage bias in the genome (Jia and Higgs 2008; Hershberg and Petrov 2008; Chen et al. 2014). The correlation between preferred codons and the frequency of the corresponding tRNA has been shown (Xia 2005) since tRNA is involved in protein translation. Translational selection occurs when one codon pairs more efficiently than another with the anticodon arm of the corresponding tRNA (Zhang et al. 2017). Translational selection acting on mtDNA may not act towards tRNA gene numbers, unlike nuclear DNA, since the number of available tRNAs is limited to 1 for each amino acid, except for leucine and serine (two types of tRNA to mtDNA) (Hershberg and Petrov 2008).
The frequency of codon usage in mitochondrial proteins is related to the positions of tRNA along the mtDNA (for example, the codons of tRNA near the control region where transcription efficiency is high were used more frequently (Chang and Clayton 1986)). The codon CTA (Leu) was selected versus TTA (Leu) (which was closer to the control region) for leucine and AGC (Ser) versus TCA (Ser) for serine (only Ser and Leu have two tRNAs encoded in the mitogenome) in the protein-coding genes of Clupeoids, indicating the translation efficiency-associated limitation acting on mtDNA codon usage (Satoh et al. 2010). Thus, the codon usage pattern in Clupeoids favors efficient translation. In addition, hydrophobic amino acids, which are abundantly used in the synthesis of the mitochondrial membrane protein complex, were preferred (Satoh et al. 2010). The exceptional use of Methionine (with anticodon 5’-CAT-3’instead of TAT, frequent codon ATA) and Proline (with anticodon 5’-AGG-3’instead of GGG, frequent codon CCT) codon/anticodon, deviating from the common codon usage bias may be related to the predominant role of selection associated with translational initiation indicating that the translation initiation rate is more important than elongation (Xia 2005).
Mutation pressure, anticodon sites and tRNA position
Either strand-specific mutational bias (mutation hypothesis) or selection in codon-anticodon adaptation (selection hypothesis) have been proposed as two possible mechanisms shaping the anticodon of tRNAs (Xia 2005; Satoh et al. 2010). We found that the anticodons of all tRNAs, regardless of their source strand (i.e., both L and H strands), are saturated with the maximum possible G/T substitutions within the constraints of the vertebrate codon table. A gradient also exists in the position of the tRNA between OL, OH and the control region based on the GT content in their anticodon sites. Both these observations negate the strand-specific mutation bias model (mutation hypothesis) as the possible mechanisms shaping the anticodons, but this may be a pattern involved as an adaptation to deamination mutation pressure. At the same time, the anticodon also follows the selection hypothesis of anticodon versatility i.e., for two-fold degenerate codon families ending with C or U, the apparent anticodon wobble site will be G because G pairs with both C and U. Whereas, for two-fold degenerate codons ending with A and G and fourfold degenerate codons a wobble U will give the anticodon more versatility than other nucleotides (Xia 2005). This hypothesis was consistent with our result, except for tRNA Met and Pro.
Deamination commonly occurs in single-stranded DNA exposed during replication or transcription, resulting in A- to-G and C-to-T mutations on the H-strand, making the H-strand richer in G and T and consequent mutations in L-strand accumulate A and C (Shadeland Clayton 1997; Lowell and Spiegelman 2000). Due to the displacement mode of mtDNA replication, there is a gradient in deamination pressure along the direction of L-strand replication (lower to higher) between OH-OL and OL-OH of vertebrate mtDNA (Xia 2019). The evolutionary adaptation of Clupeoids to this deamination-induced mutational pressure was evident from the GT saturation of anti-codon sites (other than tRNA Met and Pro) and their logical ordering along the mtDNA. Using tRNAs from high GT anticodon sites from each anticodon family and arranging them according to GT content along the mtDNA, colinear with the deamination pressure exhibited between OL and OH, can protect tRNA anticodons from further mutation by deamination (Satoh et al. 2010). Furthermore, Clupeoids retained a codon usage bias in the protein-coding region with a strong anti-G bias and codon abundance with A and C at the 3rd codon position compared to those with T. The frequency of codon usage in mitochondrial proteins is related to the positions of tRNA along mtDNA (i.e., codons of tRNA near the control region were heavily used). Therefore, the protein-coding region of Clupeoid mitogenomes evolved into a codon usage pattern in which most of them are complementary to the GT-saturated tRNA anticodons in the mitogenome. This observation disproved the codon-anticodon adaptation (selection) hypothesis that codon usage bias is maintained by strand-specific mutational bias and that biased codon usage drives anticodon evolution (Xia 2005; Satoh et al. 2010). From this, it can be concluded that the deamination-related pressure may be stronger than the codon/amino acid-related constraints in the vertebrate mitogenome, thus affecting the anticodon composition and the order/position of the tRNA in the genome. The codon usage pattern was evolved towards high translational efficiency (codon/amino acid-related constraints), as evidenced by a pattern in which most of them are complementary to the GT-saturated tRNA anticodon sites (maintained by deamination-related pressure) in their mitogenome and the usage of codons of the tRNA genes located near the control region (fixed by deamination pressure), where transcription efficiency was high.
The shift in codon usage between habitats; Marine and Euryhaline/Freshwater Clupeoids
The observed codon usage bias in mtDNA was created and maintained by the result of a balance between two forces: mutational bias created by deamination mutation during their replication (in addition to natural mutation and genetic drift) and selection for translational optimization to meet the energy needs (to maintain optimal physiological process) of organisms in their habitat. Otherwise, the codons could have been fixed in the Clupeoid mtDNA (genes), since there is only one type of anticodon in each mitochondrial-encoded tRNA, available for the synthesis of most amino acids in the mitochondrial-encoded proteins (but the start codon AUG is one exception as it is necessary for efficient translation initiation). Codon preference in a gene results from a balance between mutational bias and natural selection to optimize translation (Rand and Kann 1998). Relatively high expression of a gene is associated with a full pairing of its codons with corresponding tRNA anticodons and tRNA abundance (Xia 2005). Acclimatization of marine fish to euryhaline and freshwater has been demonstrated with a characteristic increased mitochondrial gene expression/protein production (Hwang and Lee 2007; Lam et al. 2014; Zhang et al. 2017). Our analysis revealed that the high level of RSCU, very high anti-G bias, and A affinity at 3rd codon position in the freshwater and euryhaline lineages correlated well with high energy demand expected for fish in euryhaline and freshwater systems. Thus, the observed difference in codon preference in marine, euryhaline, and freshwater lineages is the result of translational selection for highly expressed mitochondrial protein-coding genes necessary during the transition or migration from sea to freshwater (Whitehead et al. 2012; Hughes et al. 2017). The connection between codon usage bias and increased gene expression/protein production (translational selection) has been proven in many studies (Plotkin and Kudla 2011; de Oliveira et al. 2021; Liu et al. 2021; Zhao et al. 2021). However, due to a lack of studies, the hypothesis about the association between codon usage and habitat remains speculative until proven experimentally. The codon adaptation index value (based on Rattus norvegicus) of Clupeoid mitochondrial protein-coding genes ranged from 0.5 to 0.6, indicating a comparatively high expression level. Clupeoid fishes originated and diversified in marine habitats and so the metabolic requirement (including mitochondrial energy/ATP synthesis) and codon usage would be optimized for the energy demands of marine habitat over millions of years of evolution under various mutations and selection pressures. This would have happened before the spread/adaptation of Clupeoids to euryhaline/freshwater habitats. The high effective population size of marine Clupeoids might have contributed significantly to this optimization process. The shift in habitat characteristics might have shifted the balance between these two forces towards translational optimization, which would be necessary to meet the high energy demands of the new habitat while maintaining homeostasis. This observation is also a conformation for the balancing force (mutational bias generated by deamination mutation in addition to spontaneous mutation, genetic drift, and selection for translational optimization) that maintains codon usage bias in vertebrate mitochondrial DNA (Rand and Kann 1998; Xia 2005; Satoh et al. 2010).
Relaxed purifying selection and Positive selection
The highest number of positively selected amino acid sites and radical modifications were observed in genes ND2, ND4 and ND5, although these sites were not associated with known functional sites. The Clupeoid lineage converging to temperate waters (lineages 2 and 4) and those converging at the marine to freshwater transition had a relatively high number of radical amino acid changes and candidate sites for positive selection/unique amino acid substitutions (in ND2, ND4, ND5, and ND6). However, the candidate sites for positive selection were disproportionately concentrated in the complex I in many fishes which may be related to the less conserved protein function (Garvin et al. 2015a, 2015b; Caballero et al. 2015; Consuegra et al. 2015) indicating that complex I, which produce 40% of the proton-pumping required for ATP synthesis is under relaxed purifying selection and these selected sites can be considered as false positives in Clupeoids.
Cytochrome c oxidase (complex IV), catalyzes the final step in the mitochondrial electron transfer chain (Li et al. 2006). It is characterized by the intrinsic uncoupling property (Kadenbach 2003) that regulates coupling efficiency to produce ATP or heat, and consequently radical amino acid changes in complex IV were less. Freshwater-specific substitutions (probably of some functional importance) were recorded in the CO2 of lineages 1 and 3 (at site #44). The amino acid cysteine (C, site #44) was common to all freshwater Clupeoids in lineages 1 and 3 except P. richmondia. In contrast, this amino acid was replaced by leucine (Tenualosa), alanine (lineage 5, Engraulidae, Pristigasteridae, Dussumierriani), and serine in all other lineages except E. thoracata (possess cysteine as in freshwater lineages). P. richmondia is the Australian catadromous herring derived from a marine ancestor, which may account for the lack of this amino acid substitution. The presence of cysteine (site #44) in freshwater-adapted E. thoracata (family Engraulidae) may indicate possible reinvasion into marine or estuarine habitats along the IWP region (Lavoue et al. 2013). All of this evidence suggests that the presence of these specific substitutions may be an ancestral polymorphism rather than convergent evolution that offers an advantage during freshwater colonization. The lineages 1, 2, and 3 were formed by one of the three dispersal events crossing the K-Pg extinction boundary and subsequent allopatric cladogenesis (Lavoue et al. 2013). Adaptation to freshwater thus took place in different places and at different times. The convergence of these amino acid substitutions in CO2 may be associated with increased energy demands in a freshwater environment, indicating the role of these proteins in osmoregulatory processes.
Colonization of Clupeoids in different habitats would have created a regime of positive directional selection in multiple mitochondrial protein-encoding genes and codon usage, although functional amino acid residues were maintained by strong purification selection. The concentration of radical amino acid changes to the major base nodes (node 73, 74, 75, 76 and77) like lineage converging at tropical to temperate water transition, marine to freshwater transition, and terminal branches of anadromous (T. ilisha, A. alosa, C.cultriventris, L. grossidens) and catadromous (P. richmondia, E. fimbricata) species support the hypothesis that selective constraints in genes (physiochemical changes in OXPHOS proteins) could be related to the degree of metabolic constraints in varied habitats. The lack of correlation between the number of radical physiochemical amino acid changes (positive selection) with non-synonymous changes (genetic distance) in the internal branches reinforced this observation. Due to the lack of recombination in the mitogenome, the high level of genetic drift would have led to the rapid fixation of nucleotide variations in small ancestral populations adapted to new habitats. Such nucleotide fixation can even occur at deleterious mutation sites, leading to the generation of patterns indistinguishable from those due to positive selection (Jacobsen et al. 2016). The high number of radical amino acid changes in the internal branches could also be generated by substitution saturation in the nucleotide sequence (Philippe et al. 2011). The occurrence of the high percentage of radical physiochemical amino acid changes in the predicted region of the internal helix loop together with a moderate correlation of the number of radical physiochemical amino acid changes with non-synonymous changes (genetic distance) in the terminal branches also indicates the dominant role of the relaxed purifying selection or fixation of neutral/mildly deleterious changes through genetic drift in the evolution of Clupeoids mtDNA protein-coding genes (Jacobsen et al. 2016).
Conclusion
The epithelial cells in fish gills, mainly pavement cells (PVCs) and mitochondria-rich cells (MRCs) play a key role in fish homeostasis (Lai et al. 2015). Improved mitochondrial coupling efficiency (Brijs et al. 2017) together with a higher number of mitochondria in these cells (Schreiber and Specker 2000) helps fish adapt to different habitats. The high radical amino acid changes and lineage-specific substitutions in the Clupeoid mtDNA may indicate increased mitochondrial coupling, which offers an advantage during freshwater colonization. Furthermore, the evolution of codon usage patterns in freshwater or brackish water lineages towards improved transcription efficiency is a clear indication of increased mitochondrial function, which provides better ion and water balance during adaptation to the marine environment. This is the first empirical evidence for codons evolving to adapt to anticodons in mtDNA. This study provides molecular evidence that highlights the importance of OXPHOS gene evolution in plasticity, colonization, and adaptation to new habitats. Conclusions regarding some of the candidate sites for positive selection observed in the Clupeoid mtDNA in the present study are speculative. This requires further investigation using protein models in programs such as Alfafold (Jumper et al. 2021) to understand the impact of mutations at these sites on protein function. We emphasize the need for experimental characterization of specific mutations, codon usage patterns, and their impact on the efficiency of oxidative phosphorylation and the resulting physiological effects that will aid in predicting the response of organisms to climate change.
Data availability
All DNA sequences used in this study are from the publically available database, GenBank and accession numbers are included in the manuscript Supplementary file_1_Table S4.
References
Ballard JWO, Melvin RG, Katewa SD, Maas K (2007) Mitochondrial DNA variation is associated with measurable differences in life‐history traits and mitochondrial metabolism in Drosophila simulans. Evolution 61:1735–1747. https://doi.org/10.1111/j.1558-5646.2007.00133.x
Balloux F, Handley LJL, Jombart T, Liu H, Manica A (2009) Climate shaped the worldwide distribution of human mitochondrial DNA sequence variation. P R Soc B-Biol Sci 276:3447–3455. https://doi.org/10.1098/rspb.2009.0752
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780. https://doi.org/10.1093/nar/27.8.1767
Brijs J, Sandblom E, Sundh H, Grans A, Hinchcliffe J, Ekstrom A, Sundell K, Olsson C, Axelsson M, Pichaud N (2017) Increased mitochondrial coupling and anaerobic capacity minimizes aerobic costs of trout in the sea. Sci Rep. 7:45778. https://doi.org/10.1038/srep45778
Caballero S, Duchene S, Garavito MF, Slikas B, Baker CS (2015) Initial evidence for adaptive selection on the NADH subunit two of freshwater dolphins by analyses of mitochondrial genomes. Plos One 10:e0123543. https://doi.org/10.1371/journal.pone.0123543
Camus MF, Wolff JN, Sgro CM, Dowling DK (2017) Experimental support that natural selection has shaped the latitudinal distribution of mitochondrial haplotypes in Australian Drosophila melanogaster. Mol Biol Evol 34:2600–2612
Carapelli A, Fanciulli PP, Frati F, Leo C (2019) Mitogenomic data to study the taxonomy of Antarctic springtail species (Hexapoda: Collembola) and their adaptation to extreme environments. Polar Biolo 1:1–8. https://doi.org/10.1007/s00300-019-02466-8
Chang DD, Clayton DA (1986) Identification of primary transcriptional start sites of mouse mitochondrial DNA: accurate in vitro initiation of both heavy and light strand transcripts. Mol Cell Biol 6:1446–1453. https://doi.org/10.1128/MCB.6.5.1446
Chen H, Sun S, Norenburg JL, Sundberg P (2014) Mutation and selection cause codon usage and bias in mitochondrial genomes of ribbon worms (Nemertea). Plos One 9:e85631. https://doi.org/10.1371/journal.pone.0085631
Clayton DA (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol 7:453–478. https://doi.org/10.1146/annurev.cb.07.110191.002321
Consuegra S, John E, Verspoor E, De Leaniz CG (2015) Patterns of natural selection acting on the mitochondrial genome of a locally adapted fish species. Genet Sel Evol 47:1–10. https://doi.org/10.1186/s12711-015-0138-0
Crofts AR (2004) The cytochrome bc 1 complex: function in the context of structure. Annu Rev Physiol 66:689–733. https://doi.org/10.1146/annurev.physiol.66.032102.150251
Da Fonseca RR, Johnson WE, O’Brien SJ, Ramos MJ, Antunes A (2008) The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9:119
de Oliveira JL, Morales AC, Hurst LD, Urrutia AO, Thompson CR, Wolf JB (2021) Inferring adaptive codon preference to understand sources of selection shaping codon usage bias. Mol Biol Evol 38:3247–3266. https://doi.org/10.1093/molbev/msab099
Dowling DK, Friberg U, Lindell J (2008) Evolutionary implications of non-neutral mitochondrial genetic variation. Trends Ecol Evol 23:546–554. https://doi.org/10.1016/j.tree.2008.05.011
Egan JP, Bloom DD, Kuo CH, Hammer MP, Tongnunui P, Iglesias SP, Sheaves M, Grudpan C, Simons AM (2018) Phylogenetic analysis of trophic niche evolution reveals a latitudinal herbivory gradient in Clupeoidei (herrings, anchovies, and allies). Mol Phylogenet Evol 124:151–161. https://doi.org/10.1016/j.ympev.2018.03.011
Elbassiouny AA, Lovejoy NR, Chang BS (2020) Convergent patterns of evolution of mitochondrial oxidative phosphorylation (OXPHOS) genes in electric fishes. Philos T R Soc B 375:20190179. https://doi.org/10.1098/rstb.2019.0179
Evans DH, Piermarini PM, Choe KP (2005) The multifunctional fish gill: dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol Rev 85:97–177
Foote AD, Morin PA, Durban JW, Pitman RL, Wade P, Willerslev E, Gilbert MT, Da Fonseca RR (2011) Positive selection on the killer whale mitogenome. Biol Lett 7:116–118. https://doi.org/10.1098/rsbl.2010.0638
Ganias K (2014) Biology and ecology of sardines and anchovies. CRC Press, Boca Raton
Garvin MR, Bielawski JP, Gharrett AJ (2011) Positive Darwinian selection in the piston that powers protonpumps in complex I of the mitochondria of Pacific salmon. Plos One 6:e24127
Garvin MR, Bielawski JP, Sazanov LA, Gharrett AJ (2015a) Review and meta‐analysis of natural selection in mitochondrial complex I in metazoans. J Zool Syst Evol Res 53:1–17. https://doi.org/10.1111/jzs.12079
Garvin MR, Thorgaard GH, Narum SR (2015b) Differential expression of genes that control respiration contribute to thermal adaptation in redband trout (Oncorhynchus mykiss gairdneri). Genome Biol Evol 7:1404–1414. https://doi.org/10.1093/gbe/evv078
Gershoni M, Levin L, Ovadia O, Toiw Y, Shani N, Dadon S, Barzilai N, Bergman A, Atzmon G, Wainstein J, Tsur A (2014) Disrupting mitochondrial–nuclear coevolution affects OXPHOS complex I integrity and impacts human health. Genome Biol Evol 6:2665–2680. https://doi.org/10.1093/gbe/evu208
Hershberg R, Petrov DA (2008) Selection on codon bias. Annu Rev Genet 42:287–299. https://doi.org/10.1146/annurev.genet.42.110807.091442
Hughes LC, Somoza GM, Nguyen BN, Bernot JP, Gonzalez-Castro M, Díaz de Astarloa JM, Ortí G (2017) Transcriptomic differentiation underlying marine to freshwater transitions in the South American silversides Odontesthes argentinensis and O. bonariensis (Atheriniformes). Ecol Evol 7:5258–5268. https://doi.org/10.1002/ece3.3133
Hwang PP, Lee TH (2007) New insights into fish ion regulation and mitochondrion-rich cells. Comp Biochem Phys A 148:479–497. https://doi.org/10.1016/j.cbpa.2007.06.416
Jacobsen MW, Da Fonseca RR, Bernatchez L, Hansen MM (2016) Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.). Mol Phyl Evol 95:161–170. https://doi.org/10.1016/j.ympev.2015.11.008
Jia W, Higgs PG (2008) Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol Biol Evol 25:339–351. https://doi.org/10.1093/molbev/msm259
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zídek A, Potapenko A, Bridgland A (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Kadenbach B (2003) Intrinsic and extrinsic uncoupling of oxidative phosphorylation. BBA-Bioenerg 1604:77–94. https://doi.org/10.1016/S0005-2728(03)00027-6
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lai KP, Li JW, Gu J, Chan TF, Tse WK, Wong CK (2015) Transcriptomic analysis reveals specific osmoregulatory adaptive responses in gill mitochondria-rich cells and pavement cells of the Japanese eel. BMC Genomics 16:1072. https://doi.org/10.1186/s12864-015-2271-0
Lajbner Z, Pnini R, Camus MF, Miller J, Dowling DK (2018) Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci Rep.-UK 8:9500. https://doi.org/10.1038/s41598-018-27805-3
Lam SH, Lui EY, Li Z, Cai S, Sung WK, Mathavan S, Lam TJ, Ip YK (2014) Differential transcriptomic analyses revealed genes and signaling pathways involved in iono-osmoregulation and cellular remodeling in the gills of euryhaline Mozambique tilapia, Oreochromis mossambicus. BMC Genomics 15:921. https://doi.org/10.1186/1471-2164-15-921
Lavoue S, Konstantinidis P, Chen WJ (2014) Progress in clupeiform systematics. In: Ganias K (eds) Biology and ecology of sardines and anchovies. CRC Press, Boca Raton
Lavoue S, Miya M, Musikasinthorn P, Chen WJ, Nishida M (2013) Mitogenomic evidence for an Indo-west pacific origin of the Clupeoidei (Teleostei: Clupeiformes). Plos One 8:e56485. https://doi.org/10.1371/journal.pone.0056485
Li Y, Park JS, Deng JH, Bai Y (2006) Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr 38:283–291. https://doi.org/10.1007/s10863-006-9052-z
Liu Y, Yang Q, Zhao F (2021) Synonymous but not silent: the codon usage code for gene expression and protein folding. Annu Rev Biochem 20:375–401. https://doi.org/10.1146/annurev-biochem-071320-112701
Lowell BB, Spiegelman BM (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404.6778:652–660. https://doi.org/10.1038/35007527
McLean MJ, Wolfe KH, Devine KM (1998) Base composition skews, replication orientation and gene orientation in 12 prokaryote genomes. J Mol Evol 47:691–696. https://doi.org/10.1007/PL00006428
Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S et al. (2003) Natural selection shaped regional mtDNA variation in humans. P Natl Acad Sci USA 100:171–176
Morales HE, Pavlova A, Joseph L, Sunnucks P (2015) Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol Ecol 24:2820–2837
Morales HE, Pavlova A, Amos N, Major R, Bragg J, Kilian A, Greening C, Sunnucks P (2016) Mitochondrial nuclear interactions maintain a deep mitochondrial split in the face of nuclear gene flow. Bio Rxiv 1:095596
Mossman JA, Jennifer YG, Navarro F, Rand DM (2019) Mitochondrial DNA fitness depends on nuclear genetic background in Drosophila. G3-Genes Genom Genet 9:1175–1188. https://doi.org/10.1534/g3.119.400067
Murrell B, Joel OW, Sasha M, Thomas W, Konrad S, Pond SLK (2012) Detecting individual sites subject to episodic diversifying selection. Plos Genet 8:e1002764
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K (2013) FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Bio Evol 30:1196–1205
Perna NT, Kocher TD (1995) Patterns of nucleotide composition at four fold degenerate sites of animal mitochondrial genomes. J Mol Evol 41:353–358. https://doi.org/10.1007/BF00186547
Philippe H, Brinkmann H, Lavrov DV, Littlewood DTJ, Manuel M, Worheide G, Baurain D (2011) Resolving difficult phylogenetic questions: why more sequences are not enough. Plos Biol 9:e1000602
Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12:32–42. https://doi.org/10.1038/nrg2899
Pond SLK, Frost SD (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533. https://doi.org/10.1093/bioinformatics/bti320
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 7:1253–1256. https://doi.org/10.1093/molbev/msn083
R Core Team (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/
Rand DM, Kann LM (1998) Mutation and selection at silent and replacement sites in the evolution of animal mitochondrial DNA. Genetica 102/103:393–407. https://doi.org/10.1023/A:1017006118852
Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC (2004) Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303:223–226
Satoh TP, Sato Y, Masuyama N, Miya M, Nishida M (2010) Transfer RNA gene arrangement and codon usage in vertebrate mitochondrial genomes: a new insight into gene order conservation. BMC Genomics 11:479. https://doi.org/10.1186/1471-2164-11-479
Schreiber AM, Specker JL (2000) Metamorphosis in the summer flounder, Paralichthysdentatus: thyroidal status influences gill mitochondria-rich cells. Gen Comp Endocr 117:238–250. https://doi.org/10.1006/gcen.1999.7407
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modelling server. Nucleic Acids Res 31:3381–3385. https://doi.org/10.1093/nar/gkg520
Sebastian W, Sukumaran S, Zacharia PU, Muraleedharan KR, Dinesh Kumar PK, Gopalakrishnan A (2020) Signals of selection in the mitogenome provide insights into adaptation mechanisms in heterogeneous habitats in a widely distributed pelagic fish. Sci Rep.-UK 10:1–4. https://doi.org/10.1038/s41598-020-65905-1
Shadel GS, Clayton DA (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66:409–435. https://doi.org/10.1146/annurev.biochem.66.1.409
Shen YY, Liang L, Zhu ZH, Zhou WP, Irwin DM, Zhang YP (2010) Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc Natl Acad Sci USA 107:8666–8671. https://doi.org/10.1073/pnas.0912613107
Stier A, Bize P, Habold C, Bouillaud F, Massemin S, Criscuolo F (2014) Mitochondrial uncoupling prevents cold-induced oxidative stress: a case study using UCP1 knockout mice. J Exp Biol 217:624–630
Sueoka N (1988) Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci 85:2653–2657. https://doi.org/10.1073/pnas.85.8.2653
Teske PR, Sandoval-Castillo J, Golla TR, Emami-Khoyi A, Tine M, von der Heyden S, Beheregaray LB (2019) Thermal selection as a driver of marine ecological speciation. Proc R Soc B 286:20182023. https://doi.org/10.1098/rspb.2018.2023
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 272:1136–1144. https://doi.org/10.1126/science.272.5265.1136
Whitehead A, Roach JL, Zhang S, Galvez F (2012) Salinity-and population-dependent genome regulatory response during osmotic acclimation in the killifish (Fundulus heteroclitus) gill. J Exp Biol 215:1293–1305. https://doi.org/10.1242/jeb.062075
Woolley S, Johnson J, Smith MJ, Crandall KA, McClellan DA (2003) TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 19:671–672. https://doi.org/10.1093/bioinformatics/btg043
Wright F (1990) The ‘effective number of codons’ used in a. Gene Gene 87:23–29. https://doi.org/10.1016/0378-1119(90)90491-9
Xia X (2005) Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene 345:13–20. https://doi.org/10.1016/j.gene.2004.11.019
Xia X (2013) DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30:1720–1728. https://doi.org/10.1093/molbev/mst064
Xia X (2019) Is there a mutation gradient along vertebrate mitochondrial genome mediated by genome replication? Mitochondrion 46:30–40. https://doi.org/10.1016/j.mito.2018.06.004
Zhang X, Wen H, Wang H, Ren Y, Zhao J, Li Y (2017) RNA-Seq analysis of salinity stress-responsive transcriptome in the liver of spotted sea bass (Lateolabrax maculatus). Plos One 12:e0173238. https://doi.org/10.1371/journal.pone.0173238
Zhao F, Zhou Z, Dang Y, Na H, Adam C, Lipzen A, Ng V, Grigoriev IV, Liu Y (2021) Genome-wide role of codon usage on transcription and identification of potential regulators. Proc Natl Acad Sci USA 9:118. https://doi.org/10.1073/pnas.2022590118
Acknowledgements
We would like to thank the Director, Central Marine Fisheries Research Institute (CMFRI) and Dr P. Vijayagopal (Head, Marine Biotechnology Division, ICAR-CMFRI) for providing facilities to carry out this work. WS received a Senior Research Fellowship from the ICAR-NICRA project. This work was carried out under the institute project MBT/GEN/25, receiving funding support from the Indian Council of Agricultural Research (ICAR).
Author information
Authors and Affiliations
Contributions
WS conceived the idea, conducted the research, interpreted the results, and wrote the first manuscript. SS coordinated the research, interpreted the results and wrote the first manuscript. AG reviewed the original manuscript, provided feedback and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Bastiaan Star.
Supplementary information
Rights and permissions
About this article

Cite this article
Sebastian, W., Sukumaran, S. & Gopalakrishnan, A. Comparative mitogenomics of Clupeoid fish provides insights into the adaptive evolution of mitochondrial oxidative phosphorylation (OXPHOS) genes and codon usage in the heterogeneous habitats. Heredity 128, 236–249 (2022). https://doi.org/10.1038/s41437-022-00519-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00519-z
