
Abstract
Crop populations derived from experimental crosses enable the genetic dissection of complex traits and support modern plant breeding. Among these, multi-parent populations now play a central role. By mixing and recombining the genomes of multiple founders, multi-parent populations combine many commonly sought beneficial properties of genetic mapping populations. For example, they have high power and resolution for mapping quantitative trait loci, high genetic diversity and minimal population structure. Many multi-parent populations have been constructed in crop species, and their inbred germplasm and associated phenotypic and genotypic data serve as enduring resources. Their utility has grown from being a tool for mapping quantitative trait loci to a means of providing germplasm for breeding programmes. Genomics approaches, including de novo genome assemblies and gene annotations for the population founders, have allowed the imputation of rich sequence information into the descendent population, expanding the breadth of research and breeding applications of multi-parent populations. Here, we report recent successes from crop multi-parent populations in crops. We also propose an ideal genotypic, phenotypic and germplasm ‘package’ that multi-parent populations should feature to optimise their use as powerful community resources for crop research, development and breeding.
Similar content being viewed by others


Over recent years, numerous multi-parent populations (MPPs) have been successfully developed in crops (Huang et al. 2015; Cockram and Mackay 2018). MPPs bring together key genomic, phenotypic and germplasm resources to form a platform for research and development. In this review, we examine three themes covering new developments in crop MPP research: (1) we survey the rapidly expanding variety of crop MPPs, explaining how differences in their design and construction affect their power and precision in mapping quantitative trait loci (QTL), on which we provide a brief primer. (2) We review the use of genomic technologies in MPPs, which have proven particularly suitable for gathering dense genomic information across large populations. We make general recommendations for collecting genotypic resources in MPPs. (3) We discuss successful applications of MPPs, particularly where they have been used for breeding and pre-breeding. This includes the identification of QTL, the application of genomic prediction to MPPs, and the direct use of MPP lines as germplasm for varietal release or pre-breeding. These recent developments have shown the potential of MPPs for crop improvement.
Multi-parent populations (MPPs)
Bi-parental populations, derived from crosses between two inbred lines, have been the standard design for genetic mapping in crops. There are three key advantages to bi-parental populations: (1) the relative simplicity of their construction. Just two generations are needed for F2 (selfed/inter-crossed F1 hybrids) populations, and only about six further generations of inbreeding in self-fertilising species are needed to make recombinant inbred lines (RILs) whose genomes are fixed. (2) Their high power to detect QTL because all allele frequencies are typically close to the optimal value of 50%. (3) The low rate of linkage disequilibrium decay within chromosomes. There are normally only one or two recombinants per chromosome arm (inbreeding a RIL only adds about one observable recombinant per arm) meaning only a few hundred genotyped markers are needed to map QTL.
However, bi-parental populations have two principal disadvantages: the lack of mapping precision, which stems from limited effective recombination occurring during population development, and low genetic diversity, which is due to the genetic bottleneck caused by the choice of two founders. This may limit the number of QTL captured as no more than two alleles segregate at any locus. Consequently, around a decade ago, a second generation of experimental mapping populations, initially utilising additional crossing generations in a bi-parental but eventually inter-crossing multiple parents (MPPs), was developed to address these issues.
The limited genetic recombination in bi-parental populations was first addressed via the advanced inter-cross (AIC) design. These capture additional recombination via crossing the F2 generation for further generations prior to genetic mapping, effectively increasing the mapping precision (Dudley 1993; Darvasi and Soller 1995). Despite its potential benefits, AIC has seldom been used in crops. So far, examples of AIC exist in two plant species, thale cress (Arabidopsis thaliana, Gerald et al. 2014) and maize (Zea mays, Lee et al. 2002; Balint-Kurti et al. 2010), discussed further by Cockram and Mackay (2018). A possible reason for the lack of uptake, acknowledged by Darvasi and Soller (1995), is that simply increasing population size in bi-parental populations also increases mapping precision. Although large bi-parental populations also require increased phenotyping and genotyping, there is no requirement for additional crossing to create the population, which is particularly important for selfing species where manual crossing is onerous.
Currently, the two most popular MPP designs in plants are nested association mapping (NAM) and multi-parent advanced generation inter-cross (MAGIC) populations. NAM population construction involves a series of crosses between a recurrent founder line and a number of alternative founders (Fig. 1). NAMs can be thought of as sets of bi-parental populations all linked by a common parent. They are therefore conceptually familiar for those used to working with bi-parental populations. While NAM captures additional genetic diversity, increased genetic recombination is essentially only captured via increasing the numbers of lines screened—as is the case in bi-parental populations. In contrast, the MAGIC design is more complex. MAGIC is an extension of AIC in some respects, except several founders are inter-crossed over multiple generations before selfing to generate inbred lines. MAGIC populations typically descend from 4, 8 or 16 parents, consistent with a simple funnel breeding design (Fig. 1; Huang et al. 2015). This is however not essential, for example, the first MAGIC population in plants used 19 A. thaliana parents (Kover et al. 2009). Each MAGIC line usually inherits alleles from all parents, and MAGIC chromosomes are random mosaics of the founder haplotypes. By capturing increased genetic recombination and genetic variation, MAGIC populations are designed to address both of the principal limitations of bi-parental populations for QTL mapping.

The nested association mapping (NAM) design consists of a series of bi-parental crosses against a common founder, from which recombinant inbred lines (RILs) are typically generated through selfing and single-seed descent (represented by dashed arrows). Hundreds of RILs can be derived from each bi-parental cross. Only four crosses are shown here but this design is readily extendable to include more founders. In the multi-parent advanced generation inter-cross (MAGIC) design, a series of equally balanced crosses are made between founders before RILs are developed.
The beneficial properties of MPPs, namely high mapping power and resolution, expanded diversity compared with bi-parental populations, and their minimal population structure has increased their uptake in crop research. This increasing popularity of crop MPPs means that many now represent mature research and development tools. Most of the world’s major crops have spawned several MPPs (Table 1) and new MPPs for other crops are imminent (e.g., pigeonpea, Cajanus cajan and chickpea, Cicer arietinum, MAGIC populations with whole-genome sequence data, Pandey et al. 2016; Roorkiwal et al. 2020) or under development (e.g., sunflower, Helianthus annuus, Matias Dominguez, personal communication and Triticum uratu, MDA, personal communication). Where multiple MAGIC and/or NAM populations for the same crop are available, these are usually descended from different founders (Table 1). Availability of multiple MPPs offer the chance to replicate QTL across MPPs and to combine MPPs to improve power (Li et al. 2015).
While this review focuses on NAM and MAGIC populations, many conceivable crossing designs can recombine multiple founders. Whatever the design, MPPs are generally: (a) derived from an explicit set of founders, preferably highly inbred lines or varieties that reproduce faithfully, (b) produced through experimental crosses that minimise selection and (c) composed of a large number of recombined individuals that are analysed together as a population. Philosophically, MPPs are established for long-term durability rather than short-term expediency. Bi-parental crosses are very efficient for mapping genetic variants that are known or suspected to vary between parents. MPPs are typically made with greater agnosticism about target traits and/or causal variants. Figure 2 demonstrates that large MPPs give a relatively good chance of detecting QTL that segregate in the population from which the founders were chosen. Thus, once constructed, MPPs provide enduring platforms on which it may be worth mapping non-target traits and/or investigating the genetic architecture of complex traits and the relationships between traits in the source population more generally.

The founders are assumed to be randomly selected from a source population in which a bi-allelic causal variant segregates at a particular minor allele frequency (MAF). Detection probability reflects both the number of founders that vary at the causal locus (calculated using binomial sampling) and the corresponding mapping power within the experimental population, which was calculated using a significance threshold of 10−4 and assuming that 5% of the phenotypic variation in a bi-parental population would be explained by the QTL: \({\it{\upbeta} }_{{\it{{\mathrm{QTL}}}}}^2 = 0.2\), \({\it{\upsigma }}^2 = {\it{\upsigma }}_{{\it{{\mathrm{QTL}}}}}^2 + 0.95\) see Eq. (1).
MPP founder selection
Founder selection is a key issue in MPP design because it determines the pool of genetic variation that segregates in the population. In Fig. 2, we assume that the founders are chosen randomly from a source population. However, many MPPs specifically aim to maximise the genetic diversity captured by the founders, which can be aided by genetic algorithms for founder selection (Ladejobi et al. 2016). In addition, different MPPs may target different source populations of interest, which is typically the case when multiple MPPs exist for the same crop. For example, some MPPs target diversity among elite lines from a particular region (Bandillo et al. 2013; Mackay et al. 2014; Sannemann et al. 2018; Kidane et al. 2019), while other MPPs include landraces and wild accessions (Kover et al. 2009; Maurer et al. 2015). In other cases, founders are selected with particular traits in mind, e.g., MAGIC rice populations focussed on heat and biotic stress resistance (Leung et al. 2015). Table 1 includes brief details of founder selection strategies implemented across various MPPs to date.
Diverse, trait-specific and region-specific MPPs are useful in different scenarios. In general, those MPPs developed to maximise segregating genetic variation are rich in novel allelic combinations from the diverse founders. This makes them a permanent resource to analyse the genetic basis of complex traits in different environments. However, diverse MPPs are potentially less useful for the direct use of their germplasm in pre-breeding because undesirable alleles may segregate in the progeny (Huang et al. 2015). In contrast, MPPs based on a more conservative founder selection strategy optimised around particular varieties, traits or environments might be more quickly translated into superior breeding lines and are of greater immediate value to breeders. An intermediate MPP design may employ a mix of improved and adapted breeding lines together with diverse varieties or varieties for high yield, disease resistance and tolerance to abiotic stresses. When an MPP produces lines that combine several desirable traits, they can be used directly in breeding programmes (Descalsota et al. 2018; Zaw et al. 2019).
In the case of NAM populations, the use of a single recurrent founder that will be represented in 50% of the genomes of the resulting RILs requires particular attention. The recurrent founder may be chosen to be a standard reference variety. For example, a variety with a sequenced genome was chosen in maize (McMullen et al. 2009). If the population is designed to be used for pre-breeding material, it is key that the recurrent founder has good agronomic performance. For example, the Ethiopian durum wheat NAM was designed to mix diverse Ethiopian landraces with an elite international variety (Kidane et al. 2019). To reduce the representation of potentially maladaptive exotic genetic material in the population, backcrossing to an elite recurrent founder is also sometimes performed prior to RIL development (Jordan et al. 2011; Nice et al. 2016; Chen et al. 2019).
Crossing design
Though sharing the same overall objective of increasing genetic diversity, NAM and MAGIC populations differ in the genetic features of their constituent RILs. Each MAGIC chromosome is a random mosaic of all the founder genomes, while a NAM chromosome is a mosaic of just the two parents in its family.
The interconnected design of NAM populations allows new families to be added incrementally. Some NAM populations capture genetic diversity from up to 90 founders and may include thousands of individual RILs (McMullen et al. 2009; Maurer et al. 2015; Bouchet et al. 2017; Kidane et al. 2019). However, although the overall diversity can be large, the recurrent founder in NAM limits the haplotypic diversity within any family of RILs (Ladejobi et al. 2016). Furthermore, the benefits of increasing genetic diversity by adding founders may need to be balanced against the number of RILs developed from each cross (Gage et al. 2020; Garin et al. 2020). We also note that tailored mapping methods may be required to account for differing recombination frequencies among the NAM’s constituent bi-parental families (Li et al. 2011).
MAGIC designs require several intermating generations, proportional to the logarithm of the number of founders (Huang et al. 2015). In addition, the founders can be brought together in many different ways, where a particular order of crossing is referred to as a ‘funnel’. For example, in a population with four founders—A, B, C and D—a line derived from [(A × B) × (C × D)] has come through a different funnel to one derived from [(A × C) × (B × D)]. The number of possible funnels is often large: a population with 8 founders has 315 possible funnels if reciprocal crosses are ignored (Mackay et al. 2014). When using fully inbred founders, the genotypes of the initial two-way cross combinations are fixed, but the subsequent four-way crosses are heterozygous with unknown genetic makeup, and alleles can be lost to sampling. Thus, replicates of each crossing combination should ideally be made for all subsequent intermating generations to capture independent recombination events. However, the large number of crosses required in order to make and replicate all funnels may be a considerable challenge to population construction.
Different approaches have been employed to minimise the crossing effort required to construct MAGIC populations. Most MAGIC populations use a much reduced subset of all possible funnels (see Table 1) and derive multiple RILs from each funnel. Stadlmeier et al. (2018) found that a small number of funnels with an extra generation of inter-crossing could capture recombination as effectively as a more comprehensive MAGIC design with all possible four-way crosses. Other designs reduce the crossing effort by using a subset of founders in each funnel (Huang et al. 2011; Li et al. 2014) or exploiting several generations of random mating (Scarcelli et al. 2007; Sallam and Martsch 2015; Islam et al. 2016; Wada et al. 2017). In sorghum and bread wheat, outcrossing for random mating has been enforced using male-sterile lines (Thépot et al. 2014; Ongom and Ejeta 2018). Of potential relevance to crop MPPs, in fruit flies (Drosophila melanogaster), interconnected MAGIC populations have been developed that expand diversity while maintaining the closed MAGIC mating design (King et al. 2012). This approach could be adopted in crop MAGIC populations that share founders (e.g., Huang et al. 2012; Shah et al. 2019) to combine desirable features from both NAM and MAGIC crossing designs.
A final comment on population structure in MPPs is required. RILs derived from the same MAGIC funnel or NAM family are more closely related than those from different MAGIC funnels or NAM families. This imposes a degree of population structure which could artificially inflate the significance of QTL if not controlled. Fortunately, there are now a suite of methods that largely eliminate the effects of unequal relatedness using linear mixed models (Bradbury et al. 2007; Kang et al. 2008; Zheng et al. 2015; Broman et al. 2019). These model the phenotypic covariance between individuals in terms of their genetic covariance, which is usually computed using correlations between marker dosages (Yang et al. 2011). In any event, population structure in experimental MPPs is generally much less pronounced than it is in populations derived from pre-existing germplasm.
Power and precision in QTL mapping: a quick primer
We next summarise the key characteristics of MPP population designs, using theoretical results on power and recombination. There is a well-established general theory to calculate the power to detect a QTL segregating in any population (Lynch and Walsh 1998). Power is a function of the sample size N and the fraction \(f = \sigma _{{\mathrm{QTL}}}^2/\sigma ^2\)of the total phenotypic variance σ2 explained by the QTL, \(\sigma _{{\mathrm{QTL}}}^2\). Specifically, the power is the probability that a chi-squared distribution with one degree of freedom and non-centrality parameter Nf exceeds q0, where q0 is the quantile corresponding to the genome-wide threshold p value for the test of the null hypothesis of no association at a locus (i.e., when the non-centrality parameter is equal to zero). Thus, power only depends on Nf: if the fraction of variance explained by a QTL, f, is halved but the sample size doubled then the power is unchanged. For inbred lines, the variance explained by a bi-allelic QTL with minor allele frequency π is
where ±\(\beta _{{\mathrm{QTL}}}\) is the deviation from the mean phenotypic effect attributed to carrying either QTL allele. This formula is for inbred lines: it should be multiplied by 2 for F2 crosses. Questions of dominance do not arise in RILs because all loci should be homozygous. The formula neatly separates the underlying biological effect of an allele, \(\beta _{{\mathrm{QTL}}}\), from the effect of allele frequency, π. Assuming that an allele has the same biological effect, \(\beta _{{\mathrm{QTL}}}\), across genetic backgrounds, power is maximised when the allele frequency is half, as occurs for QTL that segregate in bi-parental populations. Reducing the minor allele frequency from 1/2 to 1/8, as occurs for alleles that are private to one founder in eight-parent MAGIC populations, will reduce power by 7/16 = 44%, so that sample size should be increased 2.3-fold to maintain power.
There is no equivalent general theory that gives a confidence interval for a QTL as a function of the sample size, QTL effect size, genetic map, etc. The most relevant result for MPPs is from Broman (2005), who derived formulae for the map expansion of MPPs assuming that 2n founders are crossed together during the creation of each RIL (n = 1 for bi-parental and NAM populations). The probability \(Q_n(r)\) that two neighbouring marker loci with recombination fraction r descend from the same founder in each RIL is
If the markers are very close together so r is small, then \(Q_n(r) \approx \left( {n + 1} \right)r\). Thus, doubling the number of founders increases the probability of a recombinant by a linear amount. We expect, other things being equal, that a confidence interval for a 2n-parent MAGIC resembles that in a two-parent population except that it is scaled by \(Q_1(r) / Q_n(r) \approx 2/(n + 1)\), which is 50% and 40% of the width of the 2-parent RIL, for 8- and 16-parent MAGIC populations, respectively. However, in practice other factors including allele frequencies and the sample size will change, and the confidence interval for a given QTL depends strongly on the local genetic map. Many QTL studies determine confidence intervals empirically using a LOD-drop approach, where the QTL extent includes all nearby markers that have p values of association that are within a set range compared with the strongest association. This has been shown to give reliable estimates of confidence intervals provided it is calibrated appropriately for each experimental population (Manichaikul et al. 2006). Empirical results also suggest that QTL effect size has an important influence on the interval size, with strong QTL better localised than weak QTL; thus, high power generally also implies shorter confidence intervals. A high density of recombinants around a weak QTL might not improve mapping resolution very much.
Both mapping power and resolution are expected to be roughly proportional to sample size. The mean total number of breakpoints between the markers with recombination fraction r in N individuals will be appoximately \(N\left( {n + 1} \right)r\), so increasing the sample size to preserve power also increases the amount of recombination and therefore the mapping resolution. In NAMs, we note that only those families in which the QTL segregates are relevant for mapping resolution, so the effective value of N for recombination will be lower. However, NAMs are frequently larger than MAGIC populations, which preserves both mapping power and resolution in practice, at the expense of phenotyping and one-time genotyping effort (Dell’Acqua et al. 2015; Anderson et al. 2018).
Application of genomics to MPPs
Next generation sequencing (NGS) platforms have revolutionised genetics by providing a genotyping technology which can in theory assay every base of an organism’s genome (Auton et al. 2015). As DNA sequencing throughput has increased, costs have dropped by several orders of magnitude (Mardis 2017). NGS-based genotyping has become a highly cost-effective and efficient agri-genomics tool in both model and non-model crop species including those with large and complex genomes (Cao et al. 2011; Cheng et al. 2019; Haberer et al. 2019; Lachagari et al. 2019). As NGS assembly algorithms and data types have improved, whole-genome sequencing (WGS) has been widely used for de novo assembly (Schatz et al. 2014; Zapata et al. 2016; Clavijo et al. 2017; IWGSC 2018; Haberer et al. 2019), as well as for re-sequencing, transcriptome sequencing (RNAseq) and epigenetic sequencing. When combined, these approaches allow experimental annotation of genes, their transcriptional variants and their transcriptional control (ENCODE Consortium 2012).
There are two main applications of NGS to MPPs. First, de novo assembly of reference genomes for each of the founders, where the development of improved DNA sequencing, algorithms and new data types now allow the construction of chromosome-level genome assemblies (Mascher et al. 2017). This permits the construction of a multiple sequence alignment of the founders, a generalisation of the pan-genome concept that focuses more on the presence/absence of the gene catalogue across the founders (Golicz et al. 2016; Gao et al. 2019), followed by a comprehensive re-annotation of the gene models in each founder. In Gan et al. (2011) it was shown that the gene models in the 19 founders of an A. thaliana MAGIC population were so divergent that a simple ‘lift-over’ of the reference annotation to each assembly caused dramatic and largely false over-prediction of deleterious mutations (e.g., premature stops and frame-shifts); RNAseq evidence showed that the effects of most of these mutations were skipped by subtle changes in splicing. Moreover, structural rearrangements revealed by de novo assembly may be significant. For example, gene expression may be perturbed by the change in regulatory context (Imprialou et al. 2017). These results demonstrate the promise of ongoing projects to produce genome assemblies for MPP founders in crop plants (e.g., https://nam-genomes.org, https://gtr.ukri.org/projects?ref=BB%2FP010741%2F1).
Second, NGS offers the ability to genotype new and existing markers. Common marker technologies such as single-nucleotide polymorphism (SNP) genotyping arrays and Kompetitive Allele Specific PCR (KASP) assays only assess known alleles at specifically designed loci and therefore tend to miss most rare variants, and even common alleles absent from the samples used to develop the assays (Burridge et al. 2018; You et al. 2018). In contrast, NGS technologies can be used to query either the whole genome (i.e., WGS) or a smaller, reproducible fraction thereof. This second strategy is referred to as reduced representation sequencing (RRS). RRS approaches include: hybridisation (e.g., exome capture, Parla et al. 2011), restriction-site-associated DNA sequencing (RAD-seq, Baird et al. 2008), double-digest RAD-seq (Peterson et al. 2012), genotyping-by-sequencing (Elshire et al. 2011) and diversity array technology-seq (Schouten et al. 2012). Whilst RRS approaches are cheap, it should be noted that sample preparation involves a greater number of steps than WGS and so introduce a higher degree of bias, which often results in missing data. In addition, the density of polymorphic markers will be lower when coding regions are targeted because they tend to be less variable.
Sparse genotyping can limit both the power and precision of QTL mapping. Equation (1) assumes that the causal variant has been genotyped, or that a surrogate marker in perfect linkage with it has. If this assumption is violated, the variance explained by the QTL is reduced by ρ2, the squared Pearson correlation between the genotyped marker and the causal variant, reducing power accordingly. Furthermore, Eq. (2) assumes that recombination breakpoints are observable. Wherever founders are identical, at least at the genotyped markers, their recombinants are invisible, limiting mapping resolution. For example, in wheat MAGIC populations, only 50–72% of the predicted recombination events appear to have been observable, possibly because the density of markers on genotyping arrays is insufficient to distinguish between founder haplotypes (Gardner et al. 2016; Stadlmeier et al. 2018). Finally, ascertainment bias in genotyping array design can make it particularly difficult to distinguish between haplotypes from diverse founders (Dell’Acqua et al. 2015).
Dense genomic information, in contrast, can identify alleles within QTL that are putatively causal. For example, Imprialou et al. (2017) used WGS to associate signatures of structural variation with phenotypic variation, finding a deletion containing three genes within a QTL for germination time in A. thaliana. Similarly, transcriptomic data can prioritise candidate genes. For example, Dell’Acqua et al. (2015) narrowed functional candidate genes in maize to those that had expression patterns in the founders consistent with the phenotypic effects that were estimated during QTL mapping.
Imputation in MPPs: the power of haplotypes
A simple mapping strategy is to genotype and phenotype an MPP and then perform association mapping to identify QTL. While this can be successful, there are advantages in exploiting the fact that RIL chromosomes are recombination mosaics of the genomes of the founders. These mosaics can be inferred from SNPs, for example using a Hidden Markov Model (Mott et al. 2000; Liu et al. 2010; Zheng et al. 2015; Broman et al. 2019). In a bi-parental cross between inbred lines, bi-allelic polymorphic markers are sufficient to identify the parental origin of each RIL genomic locus. When there are multiple haplotypes and/or founders segregating in the populations, bi-allelic markers are not completely informative anymore and the haplotypic context given by surrounding markers is used to infer the founder that contributed each genomic locus within each RIL (Mott et al. 2000).
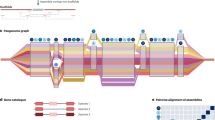
Once recombination mosaics have been constructed, sequence information from the founders can be projected onto RILs. Importantly, variants can be imputed even when they are not directly genotyped in any of the RILs. Thus, low-cost sparse genotyping data for the RILs can be used to infer the mosaics and then dense genomic data from the founders can be copied onto them to impute all variants (Fig. 3). In general, there is uncertainty in the genome mosaics and in the founder genomes, which can be easily accommodated by representing genotypes and haplotypes as dosages. Association mapping itself can be performed on the dosages of either haplotypes or imputed variants. Performing both types of association may be used to distinguish whether a QTL is caused by a bi-allelic variant or is haplotypic (Yalcin et al. 2005): evidence from an outbred eight-parent MPP in rats, Rattus rattus, suggests that ~40% of QTL are caused by multiple causal variants, or equivalently that multiple founder haplotypes have different effects (Baud et al. 2013).

a In this schematic example, the MAGIC founders are genotyped more densely and confidently than the MAGIC recombinant inbred lines. The observed genotypes in the recombinant inbred lines (white) can be used to infer the ancestry mosaics (background colours) from which unobserved genotypes (black) can be imputed. Two examples of ancestry mosaics reconstructed from low-coverage sequence data in b Arabidopsis thaliana and c bread wheat. In b, the ancestry mosaic is estimated using the Reconstruction program (http://mtweb.cs.ucl.ac.uk/mus/www/19genomes/MAGICseq.htm), and accuracy is assessed as the fraction of mismatches in each block between the inferred founder haplotypes and calls derived directly from low-coverage sequencing data. In c, the inferred ancestry proportion probabilities (dosages) are emitted after imputation using the software, STITCH (Davies et al. 2016).
Genotype imputation has been an active area of research in the past decade, and many powerful methods have been published and extensively applied already. Some of the most popular for human population studies are Beagle (Browning 2008) and IMPUTE (Howie et al. 2009). A common drawback of these established methods is that they rely on high sequencing depth and on the availability of a high-quality reference panel to infer and follow the haplotypes in the progeny. These conditions are, however, not always achieved for non-model organisms including crop species. For this reason, various methods have been developed to cater for the crop community, including mpMap (Huang et al. 2014), FILLIN (Swarts et al. 2014), STITCH (Davies et al. 2016), LB-Impute (Fragoso et al. 2016), LinkImputeR (Money et al. 2017) and magicImpute (Zheng et al. 2018). We note that the founder haplotypes of an MPP play a similar role to haplotype reference panels that are used in human genetics. However, while the latter is a construct, being simply a convenient set of basis haplotypes from which every chromosome can be derived as a mosaic, the MPP founder haplotypes are the real ancestral genomes for the MPP population and usually correspond to stable lines or varieties.
Imputation methods suggest a simple and cost-effective genotyping strategy: low-coverage (e.g., 0.3x–1.0x) WGS of each RIL. For example, using 476 A. thaliana MAGIC RILs sequenced at 0.5x, it was possible to impute SNPs with a concordance of 98% at sites previously genotyped by a conventional genotyping array (Imprialou et al. 2017). Within each constituent genome the coverage of each variable site will be random, and most sites will be covered by no more than one read. Nevertheless, by combining all the data from hundreds of RILs in a single analysis it is possible to simultaneously infer both the haplotype space and the mosaic of each chromosome in terms of these haplotypes, and hence impute the complete genome of each, even without knowledge of the founder genomes (Davies et al. 2016). It can be useful to infer founder haplotypes because unknown founders may have accidentally contributed to the population and/or the founders used for crossing may differ slightly from those sequenced. Nevertheless, the founders should ideally be characterised using orthogonal and more comprehensive genomic data; re-annotated de novo assemblies of the founders are the gold-standard. Given that researchers have to balance effort and resources, it’s significant that imputation in MPPs allows dense genomic information from a relatively small number of founder genomes to be leveraged across the population. Thus, MPPs provide one of the most cost-effective ways of obtaining near complete genomic information for a large genetically diverse population.
Applications of MPPs
A primary use of crop MPPs is to map QTL for agronomic traits to relatively narrow genetic intervals across genetically diverse backgrounds. To date, many traits have been mapped in MPPs, ranging from pathogen resistance to flavour profile. Table 2 summarises the traits mapped just in crop MAGIC populations, encompassing simple to complex traits and exemplifying their utility in dissecting the genetic architecture of crop phenotypes. For example, 75–80% of the phenotypic variance in leaf length, width and angle in a large maize NAM population can be accounted for by more than 30 QTL that have been identified (Tian et al. 2011; Gage et al. 2020).
One purpose of mapping QTL is to understand the genetic basis of traits and to identify markers that can be used in breeding programmes. In applied settings, key factors influencing whether QTL are useful are the size and stability of the effect across environments and genetic backgrounds, and the phenotyping effort required to assess the trait directly. While causal markers reduce the potential for linkage drag (introgression of linked but unfavourable alleles) during marker-assisted selection, nearby markers that tag the causal allele can be used instead. Similarly, studies of general patterns of genetic architecture do not require gene-level QTL mapping. For other theoretical and some applied applications (e.g., gene editing), validation of causal variants may be required. Thus, the research goals should generally determine the appropriate experimental design (e.g., population size and founder selection) and analysis (e.g., acceptable false positive rate), as reviewed elsewhere (Bernardo 2008, 2016).
In principle, all QTL identified from MPPs could have been mapped to the same accuracy in an appropriately chosen bi-parental population and/or a Genome-Wide Association Study (GWAS) that uses a panel of existing lines. As noted above, relative to bi-parental populations, MPPs are a more general tool within which a wider variety of traits segregate due to the increased genetic variation (Fig. 2). Compared with GWAS of pre-existing lines, experimental populations like MPPs largely avoid the potentially confounding influence of population structure and raise the allele frequency of a subset of alleles that are rare in the wider population. Thus, MPPs increase the overall probability of detecting very rare beneficial alleles. Such alleles are of particular interest for breeding (Bernardo 2016) but are difficult to detect, even in MPPs (Fig. 2). A further consideration is mapping resolution, which should be smallest in large GWAS that capture historical/natural recombination across the pre-existing germplasm, e.g., linkage decays to background levels over ~10 Kbp in wild A. thaliana accessions (Kim et al. 2007). In MAGIC populations of 527–529 RILs, QTL for various traits were mapped to intervals of 0.3–6 Mbp in A. thaliana (Kover et al. 2009) and 1.5–17 Mbp in maize (Dell’Acqua et al. 2015). In a MAGIC rice population of over 1316 RILs, the mapping intervals were ~700 Kbp on average (Raghavan et al. 2017). However, as discussed above, empirical mapping resolution may be determined by the QTL effect size and population size.
The wheat MAGIC populations developed at CSIRO (four Australian spring wheat founders, Huang et al. 2012) and NIAB (eight UK winter wheat founders, Mackay et al. 2014) are examples of MPPs that have been used to map genes controlling important yield related traits. The CSIRO spring wheat population was used to identify 18 QTL that influence the formation of paired spikelets—a modified form of inflorescence architecture in wheat. Due to the high density of polymorphic markers, the Photoperiod-1 gene could be identified as a key regulator of paired spikelet development (Boden et al. 2015). Subsequent analysis of a RIL from the same population that robustly formed paired spikelets permitted dissection of a second QTL on chromosome 4D through investigation of a RIL-specific chromosomal duplication event of 4D, which doubled the copy number of TEOSINTE BRANCHED 1 (TB1) (along with many other genes) (Dixon et al. 2018). That is, an unexpected duplication occurred during population development. The chromosomal duplication proved to be highly relevant when determining the causal gene between two closely located candidates: TB1 and the Green Revolution gene, Reduced height-1 (Rht-1). Analysis of spikelet architecture in the UK winter wheat eight-way population enabled identification of further allelic diversity for TB1. Mapping using this population demonstrated that the control of the paired spikelet formation by TB1 also occurred in winter wheat and allowed identification of a new allele of TB1 involved in the regulation of paired spikelet formation and height on chromosome 4B (TB-B1) (Dixon et al. 2018, 2020). Markers for TB1 alleles are now used for marker-assisted selection in the wheat breeding industry.
After primary mapping, QTL are often fine mapped through further crosses (Jaganathan et al. 2020). In MPPs, the intermediate crosses made during population development can be useful because, at each stage, some lines may be heterozygous for alleles at a QTL of interest. These can be used to rapidly develop pairs of near isogenic lines (NILs) differing only at one or two haplotype blocks underlying the QTL of interest. NILs and/or RILs can then be used for further molecular characterization, validating candidate genes. For example, Liller et al. (2017) used NILs to narrow down a QTL for barley awns to a region containing 66 genes and used RNA-expression data to suggest two candidate genes; Wubben et al. (2019) used virus-induced gene silencing of four candidate genes in five MAGIC RILs to identify one that was required for root-knot nematode resistance in cotton.
Separate from direct applications in breeding, QTL mapping is used by evolutionary biologists to understand the genetic basis of adaptation. For this purpose, MPPs can be used to quickly dissect the genetic basis for variation in putatively adaptive traits. For example, the A. thaliana MAGIC population has been used to fine-map several traits involved in developmental timing (e.g., time to bolting, Kover et al. 2009) and to examine the natural genetic basis of variation in seed size and number (Gnan et al. 2014). In addition to examining natural variation, MPPs could be used to identify genes under selection in experimentally evolving populations. Using wheat, for example, experimental evolution has identified natural selection for key phenology genes at contrasting geographies in France (Rhoné et al. 2008). In another application, Knapp et al. (2020) found that several genes controlling plant height and phenology reverted to the wild type in experimental wheat populations evolving under natural selection in the UK. Similar approaches could exploit the genetic variation in crop MPPs to identify additional genes underlying environmental adaptation and historical selection. For example, using MPPs with founders that span early and modern agriculture, it should be possible to examine the evolutionary history of crop improvement, identifying traits and genes involved in historical yield increases. This in turn could provide valuable insights into the potential for future yield increases.
Multi-trait analyses
Phenotyping information may be accumulated and shared in large and stable collections of inbred lines, particularly in crop MPPs. They are a convenient system in which to study interactions and correlations between traits—sometimes across multiple environments—and the extent to which their genetic basis is shared. Exploitation of MPPs to study trait–trait interactions at the level of the underlying genetics is one of their great, yet largely unfulfilled, applications. As high-throughput, high frequency phenotyping platforms become available, publicly accessible repositories of phenotypic data for multiple traits and environments will further enhance these applications.
As phenotypes for related traits are collected, multi-trait QTL analyses can be conducted (van Eeuwijk et al. 2010). Where data is incomplete, packages are available to impute missing phenotypes (Dahl et al. 2016). If multiple traits are measured, each with their own errors, but are under pleiotropic control of a single QTL, a combined analysis generalised across traits should give a more accurate indication of the true genetic effect. This has readily been applied in simple mapping populations (Hackett et al. 2001) and methods to analyse multi-trait and multi-environment traits in MPPs have been developed by Verbyla et al. (2014), allowing pleiotropic QTL and closely-linked QTL to be distinguished. Scutari et al. (2014), Descalsota et al. (2018) and Zaw et al. (2019) used Bayesian networks to simultaneously model multiple traits. Mapping composite traits derived from groups of correlated traits might uncover novel QTL that are not found when traits are analysed independently. Such analyses might suggest previously untried breeding routes to minimise trade-offs, for example, to avoid increasing yield at the expense of quality or delayed flowering, which increases the risk of encountering terminal drought (drought during grain filling) or adverse weather conditions at harvest that cause lodging or pre-harvest sprouting.
The accumulation of MPP trait data from multiple environments also enables QTL mapping of genotype by environment interactions, with the potential to breed for improved crop resilience to environmental stresses, and to understand and exploit the changes in trade-offs that occur under different environmental conditions. For example, the adaptation of A. thaliana to different environments has been studied in a MAGIC population. Several QTL were found to promote a plant’s fitness (reproductive success) in its native environment (Sweden or Italy) but reduce its fitness when transplanted to the non-native environment—a type of evolutionary trade-off (Ågren et al. 2013). In a tomato MAGIC population, trials across water, salinity and heat stress treatments revealed QTL that affect plasticity of response (Diouf et al. 2020). Garin et al. (2020) recently developed specific methods for multi-environment QTL analysis in MPPs and applied them to a European maize NAM population, finding alleles that have a greater effect on yield at sites with higher precipitation. This type of multi-environment trial analysis is also at the core of a rice MAGIC population constructed from founders that vary in their tolerance to temperature stresses (Leung et al. 2015).
Even ignoring genetic information, MPPs are valuable for dissecting correlations between phenotypes because they capture diversity and largely eliminate confounding population structure. As an illustrative example, consider plant height and root architecture in wheat. The height of wheat varieties has greatly decreased over the past century, particularly due to the introgression of large effect ‘Green Revolution’ alleles at the Rht genes that reduce the risk of yield loss through lodging (Hedden 2003). In addition, more-modern varieties tend to have smaller root systems, which may have been selected to increase yield by reducing below-ground competition within the crop (Fradgley et al. 2020). These traits (plant height and root architecture) may have a physiological connection causing them to be correlated. Alternatively, they may be correlated because modern varieties are likely to have experienced selection for both traits at independent loci. MPPs offer an opportunity to identify correlations between traits that are truly caused by a shared underlying genetic basis. For example, in a maize NAM, leaf length, width and angle have weak correlations (0.03–0.08) and share only 2–6% of QTL (Tian et al. 2011) whereas different carbon and nitrogen metabolites are more highly correlated (up to 0.7) and share QTL (Zhang et al. 2015; Gage et al. 2020).
MPP germplasm in breeding programmes
Independent of QTL analysis, MPPs provide useful germplasm for breeding or pre-breeding activities. The extensive shuffling of genetic variation during population development generates novel allelic combinations. Therefore, a subset of lines will usually display better phenotypes than any of the parental lines. These ‘transgressive’ lines may be good breeding material in their own right (Huynh et al. 2018). This is particularly true when the parents are commercial/cultivated varieties, although the MPP may have taken several years to construct since the parent varieties were released. Several MPPs employ participatory methods in the prioritization and/or selection of founders and traits (e.g., Kidane et al. 2017, 2019; Mancini et al. 2017; Campanelli et al. 2019) to facilitate end-user applications.
There are several examples where MAGIC germplasm has been used for pre-breeding or released in their own right as a variety. Li et al. (2013, 2014) report that a RIL from a rice MAGIC population was released as a new variety in China. Separate rice MAGIC populations developed at the International Rice Research Institute (IRRI) have also been used; the MAGIC RIL, IR 95099:7-B-2-10-10-2, is in the pipeline of varietal release in southern Vietnam on the basis of its maturation date, yield and grain quality, which were assessed in trials at the Cuu Long Delta Rice Research Institute, Can Tho (RKS, personal communication). In chickpea, RILs from MAGIC populations developed at the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) have been directly released as new varieties, as well as used as donors in commercial breeding programmes (RKV, personal communication). From a 16-founder wheat MAGIC population developed at NIAB, 24 MAGIC RILs have been selected for inclusion in commercial breeding programmes on the basis of their yield, ear weight and protein content (NF, personal communication). Thus, the collection of phenotypic data across several agronomically important traits in MPPs facilitates the identification of promising lines.
A major impediment to the uptake of the results of QTL studies in breeding programmes is the crossing effort required to combine beneficial alleles at multiple loci (Bernardo 2008). In large and highly recombined MPPs, RILs that ‘pyramid’ several beneficial alleles will usually already exist. For example, the rice Bio-MAGIC population developed at IRRI demonstrates the pyramiding of multiple genes for three diseases (blast, bacterial blight and brown plant hopper) without employing backcrossing (Leung et al. 2015). Furthermore, Descalsota et al. (2018) and Zaw et al. (2019) identify rice MAGIC RILs with beneficial allelic combinations across grain yield, grain zinc content, flowering time, plant height and amylose content. Four lines carry tolerant alleles for multiple diseases and insects, as confirmed by re-sequencing data. These lines are planned to be used directly in breeding programmes (Hei Leung and RKS, personal communication). In cotton, Thyssen et al. (2019) identify MAGIC RILs that pyramid eight alleles with positive effects on four different measurements of fibre quality. Thus, the identification of QTL and their carriers—often part of initial population development and analysis—also primes MPP germplasm for agricultural use.
As largely unstructured populations with associated large-scale phenotype and genotype resources, MPPs are also highly suitable for genomic prediction. This process involves fitting a model to predict phenotypes from genotypes using a training dataset. The accuracy of this prediction can then be evaluated in a test set of lines. Prediction accuracies will be most accurate when the training set and test set are highly related, and more caution should be taken when applying predictions to more distantly related material. Bian and Holland (2017) reported reasonable within-family prediction ability for plant height, southern leaf blot resistance (prediction R2 ~ 0.5) and grey leaf spot resistance (R2 ~ 0.25) in a large maize NAM. Islam et al. (2020) used a MAGIC population of cotton to predict fibre quality traits across years, finding predictive abilities (correlation coefficients) ranging from 0.41 to 0.68. Although less accurate than direct phenotyping, genomic prediction can increase genetic gain by reducing the phenotyping time and effort required for selection after the model is trained. Zhang et al. (2017) implemented this type of selection regime in an MPP generated from two rounds of inter-crossing between 18 elite tropical maize lines, finding that realised yield increased by 0.1 tonnes per hectare per year with scope for further improvement using faster genotyping protocols. A potential drawback of genomic selection is that it may select against favourable contributions from exotic founders due to linkage drag, especially in MPPs with a mixture of elite and exotic founders. For this reason, Yang et al. (2019) demonstrate the use of origin-specific genomic selection in maize and barley NAM populations, suggesting a way to maximise genetic diversity for long-term genetic gain.
The MPP ‘package’
MPPs integrate extensive genotype, phenotype and germplasm resources to provide enduring and general tools that advance theoretical knowledge and support breeding. MPP designs have allowed rich genomic information to be gathered for large populations in a cost-effective way. Furthermore, MPPs have proven to have useful applications in crop breeding. To fully realise their potential, we advocate thinking of MPPs as a package, ideally comprising:
-
(i)
The germplasm of the RILs and their founders, free of intellectual property constraints.
-
(ii)
A publicly accessible database of agronomically important phenotype data for the founders and the descendent RILs, with a system for adding further phenotypic data.
-
(iii)
Complete de novo assemblies of the founders with catalogued genetic variation (including structural variation) and gene models confirmed using RNAseq.
-
(iv)
The genome mosaics of the RILs, with founder annotations and variants projected and integrated software for mixed model GWAS at both SNP and haplotype level.
-
(v)
Demonstration that the population can be used for genomic prediction, the identification of likely causal variants underlying QTL and marker development for marker-assisted selection.
Most crop MPPs currently only have a subset of these resources available. However, the continued enrichment of MPPs with open-access phenotypic and genotypic resources will enhance their power as an enduring and growing genetic toolbox to address crop improvement.
References
Ågren J, Oakley CG, McKay JK, Lovell JT, Schemske DW (2013) Genetic mapping of adaptation reveals fitness tradeoffs in Arabidopsis thaliana. Proc Natl Acad Sci USA 110:21077–21082
Afsharyan NP, Sannemann W, Léon J, Ballvora A (2020) Effect of epistasis and environment on flowering time in barley reveals a novel flowering-delaying QTL allele. J Exp Bot 71:893–906
Anderson SL, Mahan AL, Murray SC, Klein PE (2018) Four Parent Maize (FPM) population: effects of mating designs on linkage disequilibrium and mapping quantitative traits. Plant Genome 11:1–17
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A et al. (2015) A global reference for human genetic variation. Nature 526:68–74
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:1–7
Bajgain P, Rouse MN, Tsilo TJ, Macharia GK, Bhavani S, Jin Y et al. (2016) Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS One 11:1–22
Balint-Kurti PJ, Yang J, Van Esbroeck G, Jung J, Smith ME (2010) Use of a maize advanced intercross line for mapping of QTL for Northern leaf blight resistance and multiple disease resistance. Crop Sci 50:458–466
Bandillo N, Raghavan C, Muyco PA, Sevilla MAL, Lobina IT, Dilla-Ermita CJ et al. (2013) Multi-parent advanced generation inter-cross (MAGIC) populations in rice: progress and potential for genetics research and breeding. Rice 6:1–15
Barrero JM, Cavanagh C, Verbyla KL, Tibbits JFG, Verbyla AP, Huang BE et al. (2015) Transcriptomic analysis of wheat near-isogenic lines identifies PM19-A1 and A2 as candidates for a major dormancy QTL. Genome Biol 16:1–18
Baud A, Hermsen R, Guryev V, Stridh P, Graham D, McBride MW et al. (2013) Combined sequence-based and genetic mapping analysis of complex traits in outbred rats. Nat Genet 45:767–775
Bauer E, Falque M, Walter H, Bauland C, Camisan C, Campo L et al. (2013) Intraspecific variation of recombination rate in maize. Genome Biol 14:R103
Bernardo R (2008) Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci 48:1649–1664
Bernardo R (2016) Bandwagons I, too, have known. Theor Appl Genet 129:2323–2332
Bian Y, Holland JB (2017) Enhancing genomic prediction with genome-wide association studies in multiparental maize populations. Heredity 118:585–593
Boden SA, Cavanagh C, Cullis BR, Ramm K, Greenwood J, Finnegan EJ et al. (2015) Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants 1:1–6
Bossa-Castro AM, Tekete C, Raghavan C, Delorean EE, Dereeper A, Dagno K et al. (2018) Allelic variation for broad-spectrum resistance and susceptibility to bacterial pathogens identified in a rice MAGIC population. Plant Biotechnol J 16:1559–1568
Bouchet S, Olatoye MO, Marla SR, Perumal R, Tesso T, Yu J et al. (2017) Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 206:573–585
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Broman KW (2005) The genomes of recombinant inbred lines. Genetics 169:1133–1146
Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen Ś et al. (2019) R/qtl2: software for mapping quantitative trait loci with high-dimensional data and multiparent populations. Genetics 211:495–502
Browning SR (2008) Missing data imputation and haplotype phase inference for genome-wide association studies. Hum Genet 124:439–450
Burridge AJ, Wilkinson PA, Winfield MO, Barker GLA, Allen AM, Coghill JA et al. (2018) Conversion of array-based single nucleotide polymorphic markers for use in targeted genotyping by sequencing in hexaploid wheat (Triticum aestivum). Plant Biotechnol J 16:867–876
Butrón A, Santiago R, Cao A, Samayoa RAM LF (2019) QTLs for resistance to Fusarium ear rot in a multiparent advanced generation intercross (MAGIC) maize population. Plant Dis 103:897–904
Camargo AV, Mackay I, Mott R, Han J, Doonan JH, Askew K et al. (2018) Functional mapping of quantitative trait loci (QTLS) associated with plant performance in a wheat MAGIC mapping population. Front Plant Sci 9:887
Camargo AV, Mott R, Gardner KA, Mackay IJ, Corke F, Doonan JH et al. (2016) Determining phenological patterns associated with the onset of senescence in a wheat magic mapping population. Front Plant Sci 7:1–12
Campanelli G, Sestili S, Acciarri N, Montemurro F, Palma D, Leteo F et al. (2019) Multi-parental advances generation inter-cross population, to develop organic tomato genotypes by participatory plant breeding. Agronomy 9:119
Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J et al. (2011) Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43:956–965
Causse M, Desplat N, Pascual L, Le Paslier MC, Sauvage C, Bauchet G et al. (2013) Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics 14:791
Chen Q, Yang CJ, York AM, Xue W, Daskalska LL, DeValk CA et al. (2019) TeoNAM: a nested association mapping population for domestication and agronomic trait analysis in maize. Genetics 213:1065–1078
Cheng H, Liu J, Wen J, Nie X, Xu L, Chen N et al. (2019) Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol 20:1–16
Chia JM, Song C, Bradbury PJ, Costich D, De Leon N, Doebley J et al. (2012) Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet 44:803–807
Clavijo BJ, Venturini L, Schudoma C, Accinelli GG, Kaithakottil G, Wright J et al. (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res 27:885–896
Cockram J, Mackay IJ (2018) Genetic mapping populations for conducting high-resolution trait mapping in plants. In: Varshney RK, Pandey MK, Chitikineni A (eds) Plant genetics and molecular biology. Springer, Cham, Switzerland, p 109–138
Cockram J, Scuderi A, Barber T, Furuki E, Gardner KA, Gosman N et al. (2015) Fine-mapping the wheat Snn1 locus conferring sensitivity to the Parastagonospora nodorum necrotrophic effector SnTox1 using an eight founder multiparent advanced generation inter-cross population. G3 Genes, Genomes, Genet 5:2257–2266
Corsi B, Downie RC, Venturini L, Holdgate S, Iagallo EM, Mantello CC et al. (2020) Genetic analysis of wheat sensitivity to the ToxB fungal effector from Pyrenophora tritici-repentis, the causal agent of Tan Spot. Theor Appl Genet 133:935–950
Dahl A, Iotchkova V, Baud A, Johansson S, Gyllensten U, Soranzo N et al. (2016) A multiple-phenotype imputation method for genetic studies. Nat Genet 48:466–472
Darvasi A, Soller M (1995) Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 141:1190–1207
Davies RW, Flint J, Myers S, Mott R (2016) Rapid genotype imputation from sequence without reference panels. Nat Genet 48:965–969
Dell’Acqua M, Gatti DM, Pea G, Cattonaro F, Coppens F, Magris G et al. (2015) Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays. Genome Biol 16:1–23
Descalsota GIL, Swamy BPM, Zaw H, Inabangan-Asilo MA, Amparado A, Mauleon R et al. (2018) Genome-wide association mapping in a rice magic plus population detects qtls and genes useful for biofortification. Front Plant Sci 9:1–20
Diouf I, Derivot L, Koussevitzky S, Carretero Y, Bitton F, Moreau L et al. (2020) Genetic basis of phenotypic plasticity and genotype x environment interaction in a multi-parental population. J Exp Bot eraa265. https://doi.org/10.1093/jxb/eraa265/5849330
Dixon LE, Greenwood JR, Bencivenga S, Zhang P, Cockram J, Mellers G et al. (2018) TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum). Plant Cell 30:563–581
Dixon LE, Pasquariello M, Boden SA (2020) TEOSINTE BRANCHED1 regulates height and stem internode length in bread wheat (Triticum aestivum). J Exp Bot eraa252. https://doi.org/10.1093/jxb/eraa252/5843696
Downie RC, Bouvet L, Furuki E, Gosman N, Gardner KA, Mackay IJ et al. (2018) Assessing european wheat sensitivities to Parastagonospora nodorum necrotrophic effectors and fine-mapping the Snn3-B1 locus conferring sensitivity to the effector SnTox3. Front Plant Sci 9:881
Dudley JW (1993) Molecular markers in plant improvement: manipulation of genes affecting quantitative traits. Crop Sci 33:660–668
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al. (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
ENCODE Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Fradgley N, Evans G, Biernaskie JM, Cockram J, Marr EC, Oliver AG et al. (2020) Effects of breeding history and crop management on the root architecture of wheat. Plant Soil. https://doi.org/10.1007/s11104-020-04585-2
Fragoso CA, Heffelfinger C, Zhao H, Dellaporta SL (2016) Imputing genotypes in biallelic populations from low-coverage sequence data. Genetics 202:487–495
Fragoso CA, Moreno M, Wang Z, Heffelfinger C, Arbelaez LJ, Aguirre JA et al. (2017) Genetic architecture of a rice nested association mapping population. G3 Genes, Genomes, Genet 7:1913–1926
Gage JL, Monier B, Giri A, Buckler ES, Buckler ES (2020) Ten years of the maize nested association mapping population: impact, limitations, and future directions. Plant Cell. https://doi.org/10.1105/tpc.19.00951
Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL et al. (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477:419–423
Gangurde SS, Wang H, Yaduru S, Pandey MK, Fountain JC, Chu Y et al. (2019) Nested-association mapping (NAM)-based genetic dissection uncovers candidate genes for seed and pod weights in peanut (Arachis hypogaea). Plant Biotechnol J 18:1457–1471
Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM et al. (2019) The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat Genet 51:1044–1051
Gardner KA, Wittern LM, Mackay IJ (2016) A highly recombined, high-density, eight-founder wheat MAGIC map reveals extensive segregation distortion and genomic locations of introgression segments. Plant Biotechnol J 14:1406–1417
Garin V, Malosetti M, van Eeuwijk F (2020) Multi-parent multi-environment QTL analysis: an illustration with the EU-NAM Flint population. Theor Appl Genet https://doi.org/10.1007/s00122-020-03621-0
Garin V, Wimmer V, Borchardt D, Malosetti M, van Eeuwijk F (2020) The influence of QTL allelic diversity on QTL detection in multi-parent populations: a simulation study in sugar beet. bioRxiv. https://doi.org/10.1011/2020.02.04.930677
Gerald JNF, Carlson AL, Smith E, Maloof JN, Weigel D, Chory J et al. (2014) New Arabidopsis advanced intercross recombinant inbred lines reveal female control of nonrandom mating. Plant Physiol 165:175–185
Gnan S, Priest A, Kover PX (2014) The genetic basis of natural variation in seed size and seed number and their trade-off using Arabidopsis thaliana magic lines. Genetics 198:1751–1758
Golicz AA, Bayer PE, Barker GC, Edger PP, Kim HR, Martinez PA et al. (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat Commun 7:1–8
Guan H, Ali F, Pan Q (2017) Dissection of recombination attributes for multiple maize populations using a common SNP assay. Front Plant Sci 8:1–12
Haberer G, Bauer E, Kamal N, Gundlach H, Fischer I, Seidel MA et al. (2019) European maize genomes unveil pan-genomic dynamics of repeats and genes. bioRxiv p.766444. https://doi.org/10.1101/766444
Hackett CA, Bradshaw JE, McNicol JW (2001) Interval mapping of quantitative trait loci in autotetraploid species. Genetics 159:1819–1832
Han Z, Hu G, Liu H, Liang F, Yang L, Zhao H et al. (2020) Bin-based genome-wide association analyses improve power and resolution in QTL mapping and identify favorable alleles from multiple parents in a four-way MAGIC rice population. Theor Appl Genet 133:59–71
Hedden P (2003) The genes of the Green Revolution. Trends Genet 19:5–9
Hemshrot A, Poets AM, Tyagi P, Lei L, Carter CK, Hirsch CN et al. (2019) Development of a multiparent population for genetic mapping and allele discovery in six-row barley. Genetics 213:595–613
Higgins RH, Thurber CS, Assaranurak I, Brown PJ (2014) Multiparental mapping of plant height and flowering time QTL in partially isogenic Sorghum families. G3 Genes, Genomes, Genet 4:1593–1602
Holbrook CC, Isleib TG, Ozias-Akins P, Chu Y, Knapp SJ, Tillman B et al. (2013) Development and phenotyping of recombinant inbred line (RIL) populations for peanut (Arachis hypogaea). Peanut Sci 40:89–94
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics 5:e1000529
Hu J, Guo C, Wang B, Ye J, Liu M, Wu Z et al. (2018) Genetic properties of a nested association mapping population constructed with semi-winter and spring oilseed rapes. Front Plant Sci 871:1–14
Huang BE, George AW, Forrest KL, Kilian A, Hayden MJ, Morell MK et al. (2012) A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol J 10:826–839
Huang BE, Raghavan C, Mauleon R, Broman KW, Leung H (2014) Efficient imputation of missing markers in low-coverage genotyping-by-sequencing data from multiparental crosses. Genetics 197:401–404
Huang BE, Verbyla KL, Verbyla AP, Raghavan C, Singh VK, Gaur P et al. (2015) MAGIC populations in crops: current status and future prospects. Theor Appl Genet 128:999–1017
Huang X, Paulo M-J, Boer M, Effgen S, Keizer P, Koornneef M et al. (2011) Analysis of natural allelic variation in Arabidopsis using a multiparent recombinant inbred line population. Proc Natl Acad Sci USA 108:4488–4493
Huynh BL, Ehlers JD, Huang BE, Muñoz-Amatriaín M, Lonardi S, Santos JRP et al. (2018) A multi-parent advanced generation inter-cross (MAGIC) population for genetic analysis and improvement of cowpea (Vigna unguiculata L. Walp.). Plant J 93:1129–1142
Imprialou M, Kahles A, Steffen JG, Osborne EJ, Gan X, Lempe J et al. (2017) Genomic rearrangements in Arabidopsis considered as quantitative traits. Genetics 205:1425–1441
Islam MS, Fang DD, Jenkins JN, Guo J, McCarty JC, Jones DC (2020) Evaluation of genomic selection methods for predicting fiber quality traits in Upland cotton. Mol Genet Genom 295:67–79
Islam MS, Thyssen GN, Jenkins JN, Zeng L, Delhom CD, McCarty JC et al. (2016) A MAGIC population-based genome-wide association study reveals functional association of GhRBB1_A07 gene with superior fiber quality in cotton. BMC Genom 17:1–17
IWGSC (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191
Jaganathan D, Bohra A, Thudi M, Varshney RK (2020) Fine mapping and gene cloning in the post-NGS era: advances and prospects. Theor Appl Genet 133:1791–1810
Jiménez-Galindo JC, Malvar RA, Butrón A, Santiago R, Samayoa LF, Caicedo M et al. (2019) Mapping of resistance to corn borers in a MAGIC population of maize. BMC Plant Biol 19:1–17
Jordan DR, Mace ES, Cruickshank AW, Hunt CH, Henzell RG (2011) Exploring and exploiting genetic variation from unadapted sorghum germplasm in a breeding program. Crop Sci 51:1444–1457
Jordan KW, Wang S, He F, Chao S, Lun Y, Paux E et al. (2018) The genetic architecture of genome-wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J 95:1039–1054
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ et al. (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723
Khazaei H, Stoddard FL, Purves RW, Vandenberg A (2018) A multi-parent faba bean (Vicia faba L.) population for future genomic studies. Plant Genet Resour Charact Util 16:419–423
Kidane YG, Gesesse CA, Hailemariam BN, Desta EA, Mengistu DK, Fadda C et al. (2019) A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnol J 17:1380–1393
Kidane YG, Mancini C, Mengistu DK, Frascaroli E, Fadda C, Pè ME et al. (2017) Genome wide association study to identify the genetic base of smallholder farmer preferences of durum wheat traits. Front Plant Sci 8:1–11
Kim HJ, Delhom CD, Fang DD, Zeng L, Jenkins JN, McCarty JC et al. (2020) Application of the cottonscope for determining fiber maturity and fineness of an upland cotton MAGIC population. Crop Sci https://doi.org/10.1002/csc2.20197
Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S et al. (2007) Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet 39:1151–1155
King EG, Macdonald SJ, Long AD (2012) Properties and power of the Drosophila synthetic population resource for the routine dissection of complex traits. Genetics 191:935–949
Knapp S, Döring TF, Jones HE, Snape J, Wingen LU (2020) Natural selection towards wild-type in composite cross populations of winter wheat. Front Plant Sci 10:1757
Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD et al. (2009) A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. Plos Genet 5:e1000551
Lachagari VBR, Gupta R, Lekkala SP, Mahadevan L, Kuriakose B, Chakravartty N et al. (2019) Whole genome sequencing and comparative genomic analysis reveal allelic variations unique to a purple colored rice landrace (Oryza sativa ssp. Indica cv. Purpleputtu). Front Plant Sci 10:1–15
Ladejobi O, Elderfield J, Gardner KA, Gaynor RC, Hickey J, Hibberd JM et al. (2016) Maximizing the potential of multi-parental crop populations. Appl Transl Genomics 11:9–17
Lee M, Sharopova N, Beavis WD, Grant D, Katt M, Blair D et al. (2002) Expanding the genetic map of maize with the intermated B73 x Mo17 (IBM) population. Plant Mol Biol 48:453–461
Leung H, Raghavan C, Zhou B, Oliva R, Choi IR, Lacorte V et al. (2015) Allele mining and enhanced genetic recombination for rice breeding. Rice 8:1–11
Li C, Li Y, Bradbury PJ, Wu X, Shi Y, Song Y et al. (2015) Construction of high-quality recombination maps with low-coverage genomic sequencing for joint linkage analysis in maize. BMC Biol 13:1–12
Li H, Bradbury P, Ersoz E, Buckler ES, Wang J (2011) Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS ONE, 6:e17573
Li XF, Liu ZX, Lu DB, Liu YZ, Mao XX, Li ZX et al. (2013) Development and evaluation of multi-genotype varieties of rice derived from MAGIC lines. Euphytica 192:77–86
Li Z, Ye G, Yang M, Liu Z, Lu D, Mao X et al. (2014) Genetic characterization of a multiparent recombinant inbred line of rice population. Res Crop 15:28–37
Liller CB, Walla A, Boer MP, Hedley P, Macaulay M, Effgen S et al. (2017) Fine mapping of a major QTL for awn length in barley using a multiparent mapping population. Theor Appl Genet 130:269–281
Lin M, Corsi B, Ficke A, Tan K-C, Cockram J, Lillemo M (2020) Genetic mapping using a wheat multi-founder population reveals a locus on chromosome 2A controlling resistance to both leaf blotch and glume blotch caused by the necrotrophic fungal pathogen Parastogonospora nodorum. Theor Appl Genet 133:785–808
Liu EY, Zhang Q, McMillan L, de Villena FPM, Wang W (2010) Efficient genome ancestry inference in complex pedigrees with inbreeding. Bioinformatics 26:199–207
Lopez-Malvar A, Butron A, Malvar A (2020) Genomics of maize stover yield and saccharification efficiency using a multi-parent advanced generation intercross (MAGIC) population. Preprint at https://doi.org/10.21203/rs.3.rs-16878/v1
Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer, Sunderland, MA
Mace ES, Hunt CH, Jordan DR (2013) Supermodels: Sorghum and maize provide mutual insight into the genetics of flowering time. Theor Appl Genet 126:1377–1395
Mackay IJ, Bansept-Basler P, Bentley AR, Cockram J, Gosman N, Greenland AJ et al. (2014) An eight-parent multiparent advanced generation inter-cross population for winter-sown wheat: creation, properties, and validation. G3 Genes, Genomes, Genet 4:1603–1610
Mahan AL, Murray SC, Klein PE (2018) Four-parent maize (FPM) population: development and phenotypic characterization. Crop Sci 58:1106–1117
Mancini C, Kidane YG, Mengistu DK, Pè ME, Fadda C, Dell’Acqua M et al. (2017) Joining smallholder farmers’ traditional knowledge with metric traits to select better varieties of Ethiopian wheat. Sci Rep 7:1–11
Manichaikul A, Dupuis J, Sen Ś, Broman KW (2006) Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics 174:481–489
Mardis ER (2017) DNA sequencing technologies: 2006–2016. Nat Protoc 12:213–218
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T et al. (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433
Mathew B, Léon J, Sannemann W, Sillanpää MJ (2018) Detection of epistasis for flowering time using bayesian multilocus estimation in a barley MAGIC population. Genetics 208:525–536
Maurer A, Draba V, Jiang Y, Schnaithmann F, Sharma R, Schumann E et al. (2015) Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genom 16:290
McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q et al. (2009) Genetic properties of the maize nested association mapping population. Science 325:737–740
Meng L, Guo L, Ponce K, Zhao X, Ye G (2016) Characterization of three indica rice multiparent advanced generation intercross (MAGIC) populations for quantitative trait loci identification. Plant. Genome 9:1–14
Meng L, Zhao X, Ponce K, Ye G, Leung H (2016) QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines. F Crop Res 189:19–42
Milner SG, Maccaferri M, Huang BE, Mantovani P, Massi A, Frascaroli E et al. (2016) A multiparental cross population for mapping QTL for agronomic traits in durum wheat (Triticum turgidum ssp. durum). Plant Biotechnol J 14:735–748
Money D, Migicovsky Z, Gardner K, Myles S (2017) LinkImputeR: user-guided genotype calling and imputation for non-model organisms. BMC Genom 18:1–12
Mott R, Talbot CJJ, Turri MGG, Collins ACC, Flint J (2000) A method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci USA 97:12649–12654
Naoumkina M, Thyssen GN, Fang DD, Jenkins JN, McCarty JC, Florane CB (2019) Genetic and transcriptomic dissection of the fiber length trait from a cotton (Gossypium hirsutum L.) MAGIC population. BMC Genom 20:1–14.
Nice LM, Steffenson BJ, Brown-Guedira GL, Akhunov ED, Liu C, Kono TJY et al. (2016) Development and genetic characterization of an advanced backcross-nested association mapping (AB-NAM) population of wild × cultivated barley. Genetics 203:1453–1467
Ogawa D, Nonoue Y, Tsunematsu H, Kanno N, Yamamoto T, Yonemaru JI (2018) Discovery of QTL alleles for grain shape in the Japan-MAGIC rice population using haplotype information. G3 Genes, Genomes, Genet 8:3559–3565
Ogawa D, Yamamoto E, Ohtani T, Kanno N, Tsunematsu H, Nonoue Y et al. (2018) Haplotype-based allele mining in the Japan-MAGIC rice population. Sci Rep 8:1–11
Ongom PO, Ejeta G (2018) Mating design and genetic structure of a multi-parent advanced generation intercross (MAGIC) population of sorghum (Sorghum bicolor (L.) moench). G3 Genes, Genomes, Genet 8:331–341
Pandey MK, Roorkiwal M, Singh VK, Ramalingam A, Kudapa H, Thudi M et al. (2016) Emerging genomic tools for legume breeding: current status and future prospects. Front Plant Sci 7:1–18
Parla JS, Iossifov I, Grabill I, Spector MS, Kramer M, McCombie WR (2011) A comparative analysis of exome capture. Genome Biol (9):R97
Pascual L, Desplat N, Huang BE, Desgroux A, Bruguier L, Bouchet JP et al. (2015) Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol J 13:565–577
Peiffer JA, Romay MC, Gore MA, Flint-Garcia SA, Zhang Z, Millard MJ et al. (2014) The genetic architecture of maize height. Genetics 196:1337–1356
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135
Ponce K, Zhang Y, Guo L, Leng Y, Ye G (2020) Genome-Wide Association Study of Grain Size Traits in Indica rice multiparent advanced generation intercross (MAGIC) population. Front Plant Sci 11:1–12
Ponce KS, Ye G, Zhao X (2018) QTL identification for cooking and eating quality in indica rice using multi-parent advanced generation intercross (MAGIC) population. Front Plant Sci 9:1–9
Raghavan C, Mauleon R, Lacorte V, Jubay M, Zaw H, Bonifacio J et al. (2017) Approaches in characterizing genetic structure and mapping in a rice multiparental population. G3 Genes, Genomes, Genet 7:1721–1730
Rebetzke GJ, Verbyla AP, Verbyla KL, Morell MK, Cavanagh CR (2014) Use of a large multiparent wheat mapping population in genomic dissection of coleoptile and seedling growth. Plant Biotechnol J 12:219–230
Rhoné B, Remoué C, Galic N, Goldringer I, Bonnin I (2008) Insight into the genetic bases of climatic adaptation in experimentally evolving wheat populations. Mol Ecol 17:930–943
Roorkiwal M, Bharadwaj C, Barmukh R, Dixit GP, Thudi M, Gaur PM et al. (2020) Integrating genomics for chickpea improvement: achievements and opportunities. Theor Appl Genet 133:1703–1720
Sallam A, Martsch R (2015) Association mapping for frost tolerance using multi-parent advanced generation inter-cross (MAGIC) population in faba bean (Vicia faba L.). Genetica 143:501–514
Sannemann W, Huang BE, Mathew B, Léon J (2015) Multi-parent advanced generation inter-cross in barley: high-resolution quantitative trait locus mapping for flowering time as a proof of concept. Mol Breed 35:86
Sannemann W, Lisker A, Maurer A, Léon J, Kazman E, Cöster H et al. (2018) Adaptive selection of founder segments and epistatic control of plant height in the MAGIC winter wheat population WM-800. BMC Genom 19:1–16
Scarcelli N, Cheverud JM, Schaal BA, Kover PX (2007) Antagonistic pleiotropic effects reduce the potential adaptive value of the FRIGIDA locus. Proc Natl Acad Sci USA 104:16986–16991
Schatz MC, Maron LG, Stein JC, Wences A, Gurtowski J, Biggers E et al. (2014) Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol 15:506
Schmutzer T, Samans B, Dyrszka E, Ulpinnis C, Weise S, Stengel D et al. (2015) Species-wide genome sequence and nucleotide polymorphisms from the model allopolyploid plant Brassica napus. Sci Data 2:1–9
Schouten HJ, van de Weg WE, Carling J, Khan SA, McKay SJ, van Kaauwen MPW et al. (2012) Diversity arrays technology (DArT) markers in apple for genetic linkage maps. Mol Breed 29:645–660
Scutari M, Howell P, Balding DJ, Mackay I (2014) Multiple quantitative trait analysis using Bayesian networks. Genetics 198:129–137
Septiani P, Lanubile A, Stagnati L, Busconi M, Nelissen H, Pè ME et al. (2019) Unravelling the genetic basis of fusarium seedling rot resistance in the MAGIC maize population: novel targets for breeding. Sci Rep 9:4–13
Shah R, Huang BE, Whan A, Newberry M, Verbyla K, Morell MK et al. (2019) The complex genetic architecture of recombination and structural variation in wheat uncovered using a large 8-founder MAGIC population. bioRxiv p.594317. https://doi.org/10.1101/594317
Song QJ, Yan L, Quigley C, Jordan BD, Fickus E, Schroeder S et al. (2017) Genetic characterization of the soybean nested association mapping population. Plant Genome 10
Stadlmeier M, Hartl L, Mohler V (2018) Usefulness of a multiparent advanced generation intercross population with a greatly reduced mating design for genetic studies in winter wheat. Front Plant Sci 871:1–12
Stadlmeier M, Jørgensen LN, Corsi B, Cockram J, Hartl L, Mohler V (2019) Genetic dissection of resistance to the three fungal plant pathogens Blumeria graminis, Zymoseptoria tritici, and Pyrenophora tritici-repentis using a multiparental winter wheat population. G3 Genes, Genomes, Genet 9:1745–1757
Swarts K, Li H, Alberto Romero Navarro J, An D, Romay MC, Hearne S et al. (2014) Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome 7:1–12
Thyssen GN, Jenkins JN, McCarty JC, Zeng L, Campbell BT, Delhom CD et al. (2019) Whole genome sequencing of a MAGIC population identified genomic loci and candidate genes for major fiber quality traits in upland cotton (Gossypium hirsutum L.). Theor Appl Genet 132:989–999
Thépot S, Restoux G, Goldringer I, Hospital F, Gouache D, Mackay I et al. (2014) Efficiently tracking selection in a multiparental population: the case of earliness in wheat. Genetics 199:609–623
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S et al. (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
van Eeuwijk FA, Bink MC, Chenu K, Chapman SC (2010) Detection and use of QTL for complex traits in multiple environments. Curr Opin Plant Biol 13:193–205
Verbyla AP, Cavanagh CR, Verbyla KL (2014) Whole-genome analysis of multienvironment or multitrait QTL in MAGIC. G3 Genes, Genomes, Genet 4:1569–1584
Wada T, Oku K, Nagano S, Isobe S, Suzuki H, Mori M et al. (2017) Development and characterization of a strawberry MAGIC population derived from crosses with six strawberry cultivars. Breed Sci 67:370–381
Wingen LU, West C, Waite ML, Collier S, Orford S, Goram R et al. (2017) Wheat landrace genome diversity. Genetics 205:1657–1676
Wubben MJ, Thyssen GN, Callahan FE, Fang DD, Deng DD, McCarty JC et al. (2019) A novel variant of Gh_D02G0276 is required for root-knot nematode resistance on chromosome 14 (D02) in Upland cotton. Theor Appl Genet 132:1425–1434
Xavier A, Jarquin D, Howard R, Ramasubramanian V, Specht JE, Graef GL et al. (2018) Genome-wide analysis of grain yield stability and environmental interactions in a multiparental soybean population. G3 Genes, Genomes, Genet 8:519–529
Yalcin B, Flint J, Mott R (2005) Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics 171:673–681
Yan W, Zhao HW, Yu K, Wang T, Khattak AN, Tian E (2020) Development of a multiparent advanced generation intercross (MAGIC) population for genetic exploitation of complex traits in Brassica juncea: glucosinolate content as an example. Plant Breeding 1–11
Yang CJ, Sharma R, Gorjanc G, Hearne S, Powell W, Mackay I (2019) Origin specific genomic selection: a simple process to optimize the favourable contribution of parents to progeny. G3 Genes, Genomes, Genet g3.401132.2020. https://doi.org/10.1534/g3.120.401132
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82
Yi Q, Malvar RA, Álvarez-Iglesias L, Ordás B, Revilla P (2020) Dissecting the genetics of cold tolerance in a multiparental maize population. Theor Appl Genet 133:503–516
You Q, Yang X, Peng Z, Xu L, Wang J (2018) Development and applications of a high throughput genotyping tool for polyploid crops: single nucleotide polymorphism (SNP) array. Front Plant Sci 9:104
Zapata L, Ding J, Willing EM, Hartwig B, Bezdan D, Jiao WB et al. (2016) Chromosome-level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc Natl Acad Sci USA 113:E4052–E4060
Zaw H, Raghavan C, Pocsedio A, Swamy BPM, Jubay ML, Singh RK et al. (2019) Exploring genetic architecture of grain yield and quality traits in a 16-way indica by japonica rice MAGIC global population. Sci Rep 9:1–11
Zhang J, Abdelraheem A, Thyssen GN, Fang DD, Jenkins JN, McCarty JC et al. (2020) Evaluation and genome-wide association study of Verticillium wilt resistance in a MAGIC population derived from intermating of eleven Upland cotton (Gossypium hirsutum) parents. Euphytica 216:1–13
Zhang N, Gibon Y, Wallace JG, Lepak N, Li P, Dedow L et al. (2015) Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol 168:575–583
Zhang X, Pérez-Rodríguez P, Burgueño J, Olsen M, Buckler E, Atlin G et al. (2017) Rapid cycling genomic selection in a multiparental tropical maize population. G3 Genes, Genomes, Genet 7:2315–2326
Zheng C, Boer MP, van Eeuwijk FA (2015) Reconstruction of genome ancestry blocks in multiparental populations. Genetics 200:1073–1087
Zheng C, Boer MP, van Eeuwijk FA (2018) Accurate genotype imputation in multiparental populations from low-coverage sequence. Genetics 210:71–82
Acknowledgements
We would like to thank all attendees of the workshop on Advances in Multi-Parent Populations Research, who contributed to the discussions that inspired this manuscript: John Baison, Chellapilla Bharadwaj, Martin Boer, Rowena Downie, Gina Garzon, Luciano Henrique Braz dos Santos, Wenhao Li, Swarmy Mallikarjuna, Maria Oszvald, Renata Pereira da Cruz, Rajiv Sharma, Narendra Pratap Singh, Shang Qian Xie, Chin Jian Yang. We particularly thank Karl Broman for helpful feedback.
Funding
JC, ARB and KAG were funded by Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/M011666/1, JB by BB/M013995/1, LED by BB/P010741/1, SAB by BB/P016855/1, RM and MFS by BB/M011585/1 and RM, MFS and OL by BB/P024726/1. LED was funded by the Rank Prize Fund’s New Lecturer Award and the United Kingdom Research and Innovation (UKRI) Future Leaders Fellowship grant MR/S031677/1. RKS was funded by the Generation Challenge Program (GCP) and IRRI core fund support. CVF was funded by ANPCyT 2017 2634, 2523 and CABANA project—BBSRC (BB/P027849/1).
Author information
Authors and Affiliations
Contributions
This review is largely a result of an MPP workshop in July 2019 arranged at NIAB by JC, OL, MFS and RM. All authors contributed to the writing and editing of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guest editor: Lindsey Compton
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scott, M.F., Ladejobi, O., Amer, S. et al. Multi-parent populations in crops: a toolbox integrating genomics and genetic mapping with breeding. Heredity 125, 396–416 (2020). https://doi.org/10.1038/s41437-020-0336-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0336-6
This article is cited by
-
Development and optimization of expected cross value for mate selection problems
Heredity (2024)
-
Deciphering the genetic basis of agronomic, yield, and nutritional traits in rice (Oryza sativa L.) using a saturated GBS-based SNP linkage map
Scientific Reports (2024)
-
Diploid Interspecific Recombinant Inbred Lines for Genetic Mapping in Potato
American Journal of Potato Research (2024)
-
Omics-driven utilization of wild relatives for empowering pre-breeding in pearl millet
Planta (2024)
-
A new winter wheat genetic resource harbors untapped diversity from synthetic hexaploid wheat
Theoretical and Applied Genetics (2024)
