
Abstract
Dwarf birch (Betula nana) has a widespread boreal distribution but has declined significantly in Britain where populations are now highly fragmented. We analyzed the genetic diversity of these fragmented populations using markers that differ in mutation rate: conventional microsatellites markers (PCR-SSRs), RADseq generated transition and transversion SNPs (RAD-SNPs), and microsatellite markers mined from RADseq reads (RAD-SSRs). We estimated the current population sizes by census and indirectly, from the linkage-disequilibrium found in the genetic surveys. The two types of estimate were highly correlated. Overall, we found genetic diversity to be only slightly lower in Britain than across a comparable area in Scandinavia where populations are large and continuous. While the ensemble of British fragments maintain diversity levels close to Scandinavian populations, individually they have drifted apart and lost diversity; particularly the smaller populations. An ABC analysis, based on coalescent models, favors demographic scenarios in which Britain maintained high levels of genetic diversity through post-glacial re-colonization. This diversity has subsequently been partitioned into population fragments that have recently lost diversity at a rate corresponding to the current population-size estimates. We conclude that the British population fragments retain sufficient genetic resources to be the basis of conservation and re-planting programmes. Use of markers with different mutation rates gives us greater confidence and insight than one marker set could have alone, and we suggest that RAD-SSRs are particularly useful as high mutation-rate marker set with a well-specified ascertainment bias, which are widely available yet often neglected in existing RAD datasets.
Similar content being viewed by others

Introduction
“In too small a population (1/4 N much greater than u and s) there is nearly complete fixation, little variation, little effect of selection and thus a static condition modified occasionally by chance fixation of new mutations leading inevitably to degeneration and extinction” (Wright 1931 p. 157) [N = population size, u = mutation rate and s = selection co-efficient]
Wright (1931) showed that chance will dominate in the evolution of genetic loci in a population whose effective size is low relative to the mutation rates and selection coefficients acting upon its loci. He predicted this would lead to the demise of those populations. This insight has been widely applied in conservation (Ellstrand and Elam 1993; Koskela et al. 2013), but genetic studies in plants seeking to show that population fragmentation leads to small, drifting, degenerating populations have given mixed results (Ellstrand and Elam 1993; Young et al. 1996), especially for trees (Kramer et al. 2008; Piotti 2009; Bacles and Jump 2011; Vranckx et al. 2012; Martins et al. 2016). This has led to suggestions that traits commonly found in tree species make them resilient to deleterious consequences of fragmentation (Lowe et al. 2015).
Our study species is dwarf birch (Betula nana), a monecious wind-pollinated, subarctic dwarf tree. Betula are generally considered self-incompatible, however, low levels of selfing has been reported in many species (Clausen 1966). Dwarf birch is nationally scarce in Britain where it appears to have declined significantly over the past centuries leading to fragmentation severity varying in space and time (Aston 1984; Wang et al. 2014). Threats include heavy browsing pressure, frequent moorland burning, potential genetic degradation, hybridization and climate change (Gilbert and Di Cosmo 2009; Zohren et al. 2016). The species is now under active conservation management on some Highland estates. By contrast in northern Scandinavia dwarf birch has an extensive range with largely continuous populations. The British and Scandinavian regions are thought to have a similar re-colonization history (Alsos et al. 2007; Eidesen et al. 2015) since the last glacial maximum (LGM) and so may provide a useful comparison.
Here, we quantify the decline of dwarf birch across Britain over the past three centuries, locate its extant populations and estimate their current census population sizes. We survey their genetic diversity with four genomic marker sets with a range of mutation rates and processes providing complementary information: conventional microsatellites amplified with PCR primers (for clarity termed here PCR-SSRs), single-nucleotide polymorphisms from RAD-sequencing reads divided into transitions (RAD-SNPti) and transversions (RAD-SNPtv), and microsatellites data-mined from RAD-sequencing reads (RAD-SSRs). Typically, indel mutations generating variation at SSR loci occur more frequently than substitution mutations that cause variation of SNPs (Li et al. 2002; Payseur and Cutter 2006) and transition SNPs mutate faster than transversions (Zhang and Gerstein 2003; Van Bers et al. 2010).
Due to their mutational mechanism, SSRs are expected to have higher rates of homoplasy, which may erode signatures of population differentiation over time (Estoup et al. 2002; Coates et al. 2009). Conversely, SNPs may lack the variation to distinguish recent demographic events (Morin et al. 2004). The different marker sets may also have contrasting ascertainment biases: PCR-based SSRs in particular, are likely to be from primers chosen to amplify loci with high heterozygosity. Another difference is that null alleles are more likely to occur in SSR surveys, affecting population genetic measures (Chapuis and Estoup 2007).
We use our data to address the conservation genetic status of dwarf birch populations in Scotland and its past demography. We compare overall levels of diversity between British and Scandinavian populations, and investigate how this is partitioned among British fragments. To investigate the relative importance of recent genetic drift in generating these patterns of diversity, we estimated the current census and effective population sizes. We use ABC analysis of coalescent models to make inferences about the historical events that may have contributed to the current distribution of genetic diversity among remaining British populations. Finally, we discuss our findings in the context of conserving genetic diversity in fragmented plant populations and the use of markers with differing mutation rates in such studies.
Materials and methods
Plant sampling
To quantify the decline of Betula nana in Britain, 2245 records from the years 1777 to 2012 were collated from national databases and conservation organizations. Historical sampling effort has varied substantially, concurrent with a hypothesized decline in B. nana. Thus, to account for this variation we also collected 23,990 records over the same period for B. pubescens, a related Birch species with a broader distribution that is considerably more abundant and with no significant recent population trends (Wang et al. 2014). Betula pubescens observations were divided into three time periods with equal numbers of records, B. nana records were then binned among these time periods to a geographical resolution of 0.1 degrees, with duplicates removed, and plotted using the ggplot package (Wickham 2009) in R software (R Development Core Team 2014) and RStudio (RStudio Team 2015). B. nana records were also assessed in Finland although recording schemes are sparse and recent. Thus, we referred to prevalence maps developed for the Vascular Plant Atlas of Finland (Lampinen and Lahti 2016), which support observations that the species is widespread and abundant.
Based on distribution records (Fig. 1 and Supplementary Fig. S1), fresh tissue samples from 1220 B. nana individuals were collected from 29 populations across the species’ extant range in Britain and ten populations from northern Scandinavia, during the summers of 2012–2014 (Fig. 2). Plants were identified based on leaf morphology and growth structure, and sampling locations were chosen to encompass a similar geographic scale in both regions. All populations in Britain that we could locate were sampled, including exhaustive sampling of several small populations for which only one or two individuals remain (e.g., SA, TD). Attempts were made to minimize resampling of clonal or cryptic individuals by maintaining where possible a minimum distance of five meters between samples. Following guidelines agreed with partner organizations, care was also taken that the material removed amounted to <5% of the living plant to avoid lasting damage (See Supp. Mat. for more information on sampling). Due to difficulties in DNA extraction from Betula leaves, tissue samples consisted of small twigs 10–20 cm long. A section of cambium tissue from each sample was removed, dehydrated and stored on silica gel for DNA extraction. The remaining plant material from each individual was dried in a press and then stored on acid-free paper for future reference.

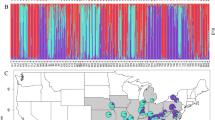
Historical records of Betula nana in Britain binned to 0.1 degree resolution, showing progressive range decline and fragmentation. Observations are normalized against records of Betula pubescens over the same period. Prevalence maps of B. nana in Finland are provided for comparison in Supplementary Fig. S1

a Sampling locations in England and Scotland (Britain). b Sampling locations in Finland and Norway (Scandinavia). c Map of Northern Europe identifying study regions. Green circles are sampled populations. Black points denote populations from historical records that could not be relocated, and thus may be locally extinct
We collected field estimates of Betula nana census population size and assigned populations to the following categories: 1–10, 10–100, 100–1000 and 1000–10,000. Where possible, at populations with ~100 or fewer individuals, we counted all individuals.
DNA extractions
Total genomic DNA was extracted from dried cambial tissue following modified cetyltrimethylammonium bromide (CTAB) protocol published in (Wang et al. 2013) and based on the approach of Doyle and Doyle (1987). The resulting DNA pellets were resuspended in 50–200 μl of TE buffer to normalize concentrations. Quality of samples was assessed using 1% gel electrophoresis and a Nanovue spectrophotometer (GE Healthcare, UK). Approximately 9% of samples were rejected due to degraded DNA, found mainly in the samples from Britain where small plant size limited the amount of tissue available. In all, 1115 DNA samples passed quality control from 39 populations. Nuclear microsatellite simple sequence repeats (PCR-SSRs) were amplified in all 1115 individuals and RAD sequencing was conducted in a subset of approximately 17% of individuals. Where samples were selected for RADseq, an additional 70% and 95% ethanol wash was added as a final stage before desiccation and rehydration in TE buffer.
PCR-SSR development and genotyping
We used both previously published (Kulju et al. 2004; Truong et al. 2005; Gürcan et al. 2010) and de novo PCR-SSR primers (Supplementary Table S1). Novel primers were developed using the v4 Dwarf birch genomic resource (http://birchgenome.org/) (Wang et al. 2013). Di-nucleotide SSR motifs were identified using a local SequenceServer BLAST (Priyam et al. 2015). Scaffolds with high hit rates were identified and then transferred into the software WEBSAT (Martins et al. 2009) to enable the identification of repeats within the scaffold and the design of primer flanking sequences. Only one new PCR-SSR assay was developed per genomic scaffold in an effort to minimize linkage of markers. Six loci were developed for the plastid genomes.
A total of 24 PCR-SSR markers were selected and tested for stutter peaks and repeatability in a subset of 12 B. nana individuals from different populations in Scotland. Combinations of useable loci were then tested in silico using the software AutoDimer (Vallone and Butler 2004) to identify possible primer dimers or cross amplification. Multiplex panels (Supplementary Table S1) were run on a 2100 Bioanalyzer (Agilent Technologies, CA, USA) to confirm expected product sizes. Fragment length was determined by auto capillary gel electrophoresis on an ABI 3730xl (Applied Biosystems). Trace peaks were called manually using the software GeneMarker® (Softgenetics, LCC) using default quality control settings. The six plastid loci (Supplementary Table S1) were found to be largely monomorphic (>0.99) and were excluded from further analysis.
The PCR-SSR data were checked for presence of null alleles, deviation from Hardy-Weinberg equilibrium (HWE) and linkage-disequilibrium (LD) using the software GENEPOP v4 (Rousset 2008), with tests adjusted for multiple comparisons using the FDR method in the base function p.adjust. Loci were tested for signatures of selection using the method of Beaumont and Nichols (1996) implemented in the software Lositan (Antao et al. 2008) over 50,000 iterations. Putative clones were identified in the software Poppr (Kamvar et al. 2014).
RAD sequencing, marker discovery, and genotyping
A subset of 190 individuals from 36 populations were selected for RAD sequencing. We used the cleavage enzyme PstI, as previous analysis of the draft B. nana genome identified an appropriate number (70,954) of cut sites (Wang et al. 2013). Cut DNA was normalized and submitted to FLORAGENEX (Portland, Oregon) for generation and sequencing of RAD tags following the protocol of Baird et al. (2008) and Hohenlohe et al. (2010). In brief, libraries were prepared with 96 unique 8-base barcodes, ligated via adapters to PstI digested genomic DNA. The resulting fragments were sequenced in two lanes of an Illumina GAIIx platform with single-end 1 × 100 bp chemistry. A single internal control sample generated from Saccharomyces bayanus was included in each lane.
Raw libraries were demultiplexed and barcode sequences checked for errors using the process_radtags module of Stacks v1.35 (Catchen et al. 2011, 2013). We used a quality threshold Phred score of ten in sliding windows of 15 bp. Three individuals failed and were removed due to low coverage. Reads were aligned to the B. nana genome and adapter sites trimmed using the software Bowtie2 (Langmead and Salzberg 2012). The mean number of retained reads per individual was 1,397,192 (Supplementary Table S2). SNP calling was performed using a bounded model in the ref_map.pl pipeline of Stacks v1.35, with two nucleotide mismatches permitted (-n 2), identified as an optimum threshold in Ilut et al. (2014) and a minimum stack depth of five (-m 5). Calls were corrected using the rxstacks module with a minimum log likelihood filter of -10 to retain a catalog locus (--lnl_lim -10) and other parameters set to default. Subsequently, cstacks and sstacks were manually rerun to rebuild and match to the catalog.
The populations module of the software Stacks (Catchen et al. 2013) was used to filter loci based on the following criteria. Loci were retained that were present in ≥70% of individuals in either of the sampling regions separately, or ≥70% of individuals across both regions combined. The final seven bases of each read were trimmed, as these showed a moderately higher frequency of SNPs possibly due to sequencing error. Putative SNPs from base 28 in all RAD loci were also removed, as the number present was highly elevated (>10×) indicating a possible error in one of the plate cycles.
For downstream demographic and Bayesian clustering analyses, polymorphisms with a minor allele frequency (MAF) of <0.01 were excluded. Similarly, to reduce LD these data were filtered to retain only a single locus per scaffold or contig in the B. nana genome assembly and achieve an r2 value of <0.2. We subsequently divided SNP loci into transition (RAD-SNPti; n = 4775) and transversion (RAD-SNPtv; n = 3306) datasets based on the type of polymorphism recorded and analyzed these independently. Finally, SNPs were exported to other analysis software using custom scripts and PGDSpider 2.1.0.1 (Lischer and Excoffier 2012).
RAD-SSR marker development
To generate RAD-SSR markers we filtered raw Illumina RADseq reads for SSR motifs by searching the consensus catalog for loci with di-nucleotide repeats (see Supplementary Information for reasoning). Initial filtering required loci to be present in >50 individuals. Reads were trimmed in the same manner as previously described and the frequency of each sequence string calculated (at least three identical reads were required for inclusion, and a minimum of ten reads per individual). Where a single sequence was present in an individual it was considered to be homozygous, where a second sequence was recorded in >10% of reads, it was called as a heterozygote (see Data Archiving for script details). Alleles were designated integer values unrelated to allele length or frequency. RAD-SSR markers were further screened for clones as described for PCR-SSR markers. For RAD-SSRs, unlike PCR-SSRs where alleles are distinguished on the basis of amplicon length, full sequence reads permitted us to distinguish alleles that differed in sequence but are the same length.
Characterizing genetic diversity
Population allelic richness (Ar) estimates were rarefied to allow correction for uneven sample size and estimated in ADZE (Szpiech et al. 2008) (Supplementary Fig. S2). Expected heterozygosity (He) was calculated across all populations and markers by region in Hierfstat (Goudet 2005) and Adegenet 2.1.1 (Jombart 2008). Population-based FIS was estimated across all markers using the method described in Catchen et al. (2013). For SSR markers, regional FIS was also co-estimated with null allele frequency using INEst (Chybicki and Burczyk 2009). Global FST was estimated by marker and region in GENEPOP (Rousset 2008).
Some statistics characterize genetic patterns that may have accumulated over long time periods, such as the local genetic diversity (heterozygosity) or differentiation among populations (FST). In order to investigate the relative importance of recent genetic drift in generating these patterns, we required an estimate of the current effective population size, (Ne), at each location. An estimate was obtained using the LD-based method of Do et al. (2014) in NeEstimator V2. This approach was cross-validated by comparison with field estimates of B. nana census population size (see above).
Finally, we report an estimate of selfing rate (David et al. 2007) and assess population structure using bayesian clustering analysis, principal component analysis, and allele frequency spectrum plots (Supplementary Materials; Figs. S3–6, S8) for each marker and region.
Population differentiation
Two different approaches were employed to assess population differentiation. In both cases we filtered for <25% missing data, genotypes in every population, and a minor allele frequency >0.05. First, pairwise FST between all population combinations was computed in Arlequin v3.5.2 (Excoffier and Lischer 2010) with significance tested over 10,000 iterations. Geographic distance matrices were calculated using Geographic Distance Matrix Generator V1.2.3 (Ersts, accessed 11-02-2016). Evidence for isolation by distance and regional discontinuities in genetic diversity were assessed using Mantel tests within fragmented and non-fragmented regions, performed in vegan (Dixon 2003). To linearize the relationship with difference, the statistic M was obtained as M = (1 – FST)/FST, and then plotted against log transformed geographic distance.
Second, to quantify the genetic divergence of each population from the regional mean, an alternative estimate of FST was defined; here termed as Maximum Likelihood FST (ML-FST). Although FST is often thought of as a statistic describing the degree of genetic differentiation among a group of sub-populations, it can also be expressed in this way, since it can be formulated as a genetic correlation within a particular subpopulation (Balding 2003). We estimated the maximum likelihood value of local FST (for a given focal population) relative to the regional mean, using the multinomial Dirichlet likelihood function proposed by Balding and Nichols (1995; example script at https://github.com/qmwugbt112/FstCalc). Finally, to evaluate the influence of sample size, we estimated ML-FST across all loci from a single individual drawn from each population.
Demographic history of dwarf birch
To infer the evolutionary and demographic history of dwarf birch in Britain since colonization, we used coalescent-based approximate Bayesian computation (ABC) implemented in DIYABC v2.0 (Cornuet et al. 2014). While there are a substantial number of possible scenarios and variables, we limited our analysis to three models addressing our core questions, as overly numerous or complex scenarios can result in poor parameter estimates (Bertorelle et al. 2010).
We performed our analysis in two steps. Initially, we used a pair of simple models, with a single contemporary British meta-population Ne variable (NeBr), to test for evidence of a historic bottleneck immediately on colonization (Scenario 2) or multiple bottlenecks after population differentiation (Scenario 1), potentially as a result of recent anthropogenic impacts. Subsequently, we incorporate census population information into the best-supported scenario (Scenario 1b), permitting independent British population Ne values to refine our posterior estimates. We test this against the null meta-population scenario from the previous analysis (Scenario 1). To further reduce the complexity of the analysis, we subsampled our full dataset (similar to the approach of Tsuda et al. 2015), retaining nine representative well-spaced populations from across the British distribution. For each marker set, this reduced dataset was tested together with an out-group formed from three Scandinavian populations from the center of our sampling distribution.
In all cases we generated 106 simulations for each scenario across each marker set. Choice of summary statistics differed across marker sets largely due to their inherent characteristics; for example RAD-SSRs are incompatible with allele length or variance statistics due to the fact they are coded as unique integers. Summary statistics together with prior parameters are reported in Supplementary Tables S3–5. To evaluate our results, we checked that our observed data fell within the range of simulated datasets using principal component analysis. Once satisfied, the posterior probabilities of scenarios were estimated using logistic regression on the 1% of simulated datasets closest to the observed data. The robustness of model choice was further assessed by testing whether the observed data fell within 1000 simulated posterior datasets. Similarly, the bias and precision of parameter estimates were assessed with 1000 pseudo-observed test datasets drawn from posterior distributions, from which the difference between each point estimate and the true value was calculated. Finally, posterior parameter estimation was performed for the best-supported scenario on the 1% of closest datasets, and the information combined across marker sets into a single density distribution.
Results
Historical records showed fragmentation and decline in the range of B. nana in the Britain over recent decades (Fig. 1). By contrast, in Scandinavia B. nana occurs abundantly in continuous populations (Supplementary Fig. S1). A total of 29 populations encompassing the extant British range and 10 Scandinavian populations were sampled (Fig. 2). In Britain it is notable that several populations that can be found in historical records, particularly those at the Southern range edge, could not be relocated during our fieldwork. Several that could be relocated had a total population size of five or fewer individuals. After quality control of DNA extractions, 1115 individuals were genotyped at 24 PCR-SSR loci, and a subset 190 individuals were retained for RADseq analyses (Table 1). Forty-nine individuals from the PCR-SSR dataset (4.4%) were subsequently identified as clonal genotypes and removed.
PCR-SSR marker validation
The number of PCR-SSR alleles per nuclear locus ranged from two to 42, with 446 alleles in total (Supplementary Table S1). Four loci were found to deviate consistently from HWE across populations, most likely due to null alleles. Of these, two were also identified as putatively under positive selection, thus all were excluded from analysis of genetic differentiation.
RAD-SNP discovery
A total of 339,291,129 reads were generated from two single-end Illumina HiSeq lanes, of which 81.66% passed strict quality controls. Three individuals were removed due to low sequencing coverage. The number of reads retained per individual ranged from 373,727 to 2,852,627, with a mean of 1,397,192 (Table S2). Across all individuals an average of 78.3% of reads aligned to the B. nana reference genome. Percentage alignment was consistently higher in Scandinavian samples, despite the reference genome being of Scottish provenance. We attribute this to poorer quality DNA derived from extractions on Scottish samples, due to the smaller size of twigs and difficulties in removing all bark from cambium tissue. Despite approximately 20% of reads that did not align being excluded, the reference-mapped pipeline identified approximately a third more usable loci than when the analysis was undertaken de novo (data not shown). This is perhaps due to reference-based mapping performing better at distinguishing paralogs. A threshold of 70% presence resulted in 6092 loci containing 23,882 SNPs. Quality filtering retained 17,694 SNPs with an overall Transition/Transversion ratio of (11:10). With minor alleles occurring at a frequency of <0.01 removed, 4208 loci and 8081 SNPs were retained, of which 4775 were transition and 3306 transversion polymorphisms (144:100); thus proportionately more of the low frequency polymorphisms were transitions.
RAD-SSR discovery
Simple sequence repeat motifs were initially identified in 3507 RADseq loci, with 196 loci meeting initial filtering criteria. We excluded three further loci with >40 unique genotypes due to the risk of a high number of sequencing errors in these reads. The number of alleles per locus ranged from 2 to 31 with a total of 1758 alleles across all populations. No evidence of clones was found in any of the RADseq samples.
Marker comparison
The number of alleles across all samples ranged from 414 in PCR-SSRs to 9428 in RAD-SNPti. Data are summarized by marker and region in Table 2. Despite fewer loci, PCR-SSRs reported a greater number of alleles per locus than RAD-SSRs. Across all datasets, patterns of allelic richness (Ar) and expected heterozygosity (He) displayed a similar pattern (Table 2), being highest in the fastest mutating markers. Inbreeding estimates (FIS) were lowest in SNPs datasets, and higher among SSR data, with RAD-SSR data reporting the highest values. As expected, SSR marker FIS values were lower when co-estimated with null allele frequency.
Regional patterns of genetic diversity
Summary statistics comparing genetic diversity among the British and Scandinavian populations are shown in Table 2 (see also Supplementary Figs. S3 and S7). The five smallest British populations are excluded from these measures as they only had one or two individuals. Allelic richness and expected heterozygosity was significantly higher in Scandinavia than in Britain for all markers (Supplementary Table S6; Two sample t-test, all p ≤ 0.01), with the exception of PCR-SSR He, which displayed the opposite trend. Fixation indices were higher in Scandinavia than in Britain for both RAD-SNP datasets (Two sample t-test, all p < 0.01). For SSR markers, fixation indices were not significantly different in Scandinavia and Britain, and null alleles affected fixation index estimates more in Scandinavia than in Britain, likely because our markers were first developed using British populations. Global FST was significantly higher in Britain across all RAD markers, with 95% confidence intervals only weakly overlapping in PCR-SSR markers. The rather small difference in overall levels of genetic diversity in Scandinavia and Britain is reflected in the diversity data for individual populations (Supplementary Table S6 and Supplementary Fig. S7). Larger British populations had similar levels of genetic diversity within them as Scandinavian populations.
Among both British and Scandinavian populations, LD-based estimates of effective population size were similar to census population sizes and showed a highly significant relationship (F1,33 = 37.12, p < 0.0001; Fig. 3; data are presented in Supplementary Table S8). Interestingly, we also find evidence for selfing in British populations (s = 0.04 [0–0.26]), but much less so in Scandinavia (s = 0.02 [0–0.17]) (see Supplementary Materials; Supplementary Table S7).

Plot of census population size vs. LD-based effective population size estimates for all populations
Population differentiation
Overall we found little evidence for isolation by distance (IBD) across markers, RAD-SSRs (Britain: r = –0.003, p = 0.46; Scandinavia: r = 0.36, p = 0.055), RAD-SNPti (Britain: r = –0.003, p = 0.06; Scandinavia: r = –0.03, p = 0.51) and RAD-SNPtv (Britain: r = 0.16, p = 0.10; Scandinavia: r = 0.002, p = 0.45), except in PCR-SSRs (Britain: r = 0.28, p = 0.009; Scandinavia: r = 0.34, p = 0.044) (Fig. 4). Furthermore, in all cases small r statistics suggest that only a very limited proportion of genetic divergence is explained by distance. To confirm that differing PCR-SSR results are not due to larger sample size, we also report mantel test results with populations subsampled to six individuals (Britain: r = 0.22, p = 0.043; Scandinavia: r = 0.05, p = 0.42).

a, b Scatter plot of linearized pairwise FST vs. pairwise geographic distance for all study populations for Britain (a) and Scandinavia (b). Weak isolation by distance (p < 0.05) was detected in Britain and Scandinavian populations using PCR-SSRs. All other relationships were non-significant. c Plot of census population size estimates and maximum likelihood FST, for each marker type, using all datasets across both regions. Noise has been added to each of the four x-axis categories to facilitate visualization
The population estimates of ML-FST were significantly higher in Britain compared to Scandinavia across all markers (Two sample t-test, all p < 0.01). This is further supported by STRUCTURE analysis, which shows a stronger pattern of population structure in Britain compared to Scandinavia (Supplementary Figs. S4–5). ML-FST values were negatively correlated with latitude in Britain (PCR-SSR: F1,27 = 13.91, p < 0.001; RAD-SSR: F1,24 = 19.37, p < 0.001; RAD-SNPti: F1,24 = 24.92, p < 0.001; RAD-SNPtv: F1,24 = 27.96, p < 0.001), but showed no significant correlation in Scandinavia (Supplementary Table S8). When ML- FST was plotted against both census and LD-based estimates of effective population size there was a strong negative relationship for all markers (Fig. 4 and Supplementary Fig. S9). We further discriminated against sample size artifacts by performing the analyses with a single individual drawn from each population, which also displayed a negative relationship (Supplementary Fig. S10). The data clearly show a strong trend for markers with higher mutations rates to exhibit lower ML-FST values.
Demographic history
Coalescent modeling in DIYABC showed all marker sets support Scenario 1 over Scenario 2 (Fig. 5 and Supplementary Fig. S11), suggesting that the losses in genetic diversity in British populations mainly occurred in recent bottlenecks in differentiated local populations, rather than upon post-glacial colonization of Britain. We then tested a scenario (1b) which allows population sizes to change at some time point post-fragmentation. In this model, we included a diffuse prior on each population’s Ne centered on our census size estimates. Both RAD-SNP marker sets supported scenario 1b over scenario 1 (Supplementary Fig. S11), but the RAD-SSRs and PCR-SSRs did not strongly support one of these models over the other.

Dendrograms showing proposed scenarios to describe the evolutionary history of dwarf birch after the last glacial maximum. Line charts show posterior demographic density distributions for scenario 1b across all marker sets independently (dashed lines) and with information combined (black lines)
We estimated combined posterior parameter distributions based on scenario 1b. These supported differentiation of British and Scandinavian lineages t2 = 789 (CI = 521–1274) generations ago, presumably shortly after colonization of Britain; an event that would have substantially limited gene flow. The estimates suggest that scandinavia maintained a somewhat larger historic Ne (NeSc = 9384, CI = 4800–9826) than Britain (NeBr = 4650, CI = 4194–9494), although their confidence intervals overlap substantially. Subsequently, British populations appear to have become differentiated very recently at t1 = 23 generations ago (CI 10–415). The Ne estimates for local British populations fitted well with our census estimates and LD Ne estimates (Table 3 and Fig. 3), a result which is not due to limitations set by the priors, with the exception that the MU estimate was close to its minimum prior value (50) for all markers (the LD Ne estimate was 715 and census estimate was up to 2000). Model checking, bias and precision estimates in these data are reported in Supplementary Table S9 and Supplementary Fig. S12.
Discussion
We generated population genomic datasets for dwarf birch populations in Britain and Scandinavia using four types of markers: 18 PCR-SSR loci with a total of 414 alleles, 193 RAD-SSR loci with 1758 alleles, 4775 RAD-SNPti biallelic loci and 3306 RAD-SNPtv biallelic loci. We successfully genotyped 1066 individuals with the PCR-SSRs and 187 individuals with the RAD-based markers. The number of loci, alleles and samples for these marker sets reflect their abundance in the genome, the dynamics of their mutation and the ease of assaying them.
Overall, the four different marker types showed similar patterns among populations and regions, despite considerable differences in marker numbers and sample sizes. Estimates of most diversity measures showed a clear pattern of PCR-SSR > RAD-SSR > RAD-SNPti > RAD-SNPtv. Figures 4 and 5 (and Supplementary Figs. S7 and S9) show that population differentiation measured by FST is higher for the SNP markers than in the PCR-SSR markers, with the RAD-SSRs being intermediate. We expected all markers to give the same FIS estimates, as nonrandom mating should affect all markers equally. However SSR based markers reported slightly higher FIS values, which can be explained by our higher estimates of null allele frequencies, particularly in Scandinavia (Table 2 and Supplementary Fig. S6).
Conservation genetic status of dwarf birch populations in Scotland
Overall, we found lower allelic richness and expected heterozygosity in Britain than in Scandinavia, and higher population differentiation. We also find a weak signal of an increased selfing rate in the UK (Supplementary Table S7). Genetic diversity was lowest in the British southern trailing range edge where effective population sizes were low (Table 2 and Fig. 3), whereas larger British populations were intermediate. In the medium-term population subdivision and differentiation may maintain genetic diversity in Britain, despite reduced Ne, until local populations become too small.
In Scandinavian populations, FST estimates are smaller and there is little evidence of isolation by distance—hence gene flow appears to be moderating the accumulation of genetic differentiation (Meirmans 2015). In Britain, FST estimates are greater and vary substantially, so it might seem surprising that there is again little evidence of isolation by distance. Instead the higher FST values particularly for Southern populations suggest that these range edge populations have become isolated and increasingly differentiated over time, while central populations show patterns of lower differentiation, which is more consistent with Scandinavia.
The greater differentiation of some populations shows that dwarf birch can be vulnerable to population fragmentation, even though it shares some attributes with larger trees that could provide resilience (Lowe et al. 2015), such as longevity and wind-dispersed pollen. These traits could help maintain genetic diversity over longer times and share it over long distances. However, our plots of FST against the current census population size (Fig. 5; also see Supplementary Fig. S9 for a plot with linkage-disequilibrium based estimates of Ne), show that differentiation is clearly related to current population sizes with a strong effect in populations currently around ten individuals. This effect is also seen in loss of allelic richness values in smaller populations (Supplementary Table S6). These results suggest that the populations have been at the current population sizes for sufficient time to affect the genetic diversity—of the order of the same number of generations as the population size. This relationship between the effective population size and census size was also seen in the ABC analysis, and is relative unusual in population genetic surveys (Frankham 1995; Vucetich et al. 1997). Part of the explanation could be in the relatively small population sizes for many populations, meaning that recent population history dominates the genetic patterns.
The history of British population structure
The combined information from the four marker datasets supported a scenario, in the ABC modeling in which the distribution of genetic diversity could be explained by the recent population size of fragmented populations, with the subdivision into smaller populations dating a long time after colonization of Britain. The RAD-SNP datasets provided more information about differences in individual population Ne, favouring different population sizes when analyzed separately.
The ABC models give estimates of the timing of events in number of generations rather than years, so their interpretation requires an estimate of generation time. Betula nana as individuals can set seed in as little as 18 months in a glasshouse (personal observation), setting a lower bound on generation time. A more central estimate can be obtained if we assume divergence occurred between the ancestral British and Scandinavian dwarf birch populations (t2 = ~789 generations) just after the last ice age 10–12,000 years ago. Dividing by the number of generations gives a generation time is 10–15 years, which is biologically plausible. This calibration would place the divergence among British populations at 230–345 years ago (t1 = ~23). That would date the ABC estimate for the time of current population subdivision to a the period when large scale anthropogenic activity including lowland agriculture had begun to shape the distribution of woody plants in Scotland (Smout et al. 2005; Godwin 1975; Tipping 1994).
The estimates for the date of reduction in current population size must be treated with more caution, since for time periods that are short compared to the mutation rate similar genetic patterns can be produced by longer times at larger population size, or shorter times at smaller. The partial confounding of these parameters explains why we did not have power to estimate different times for each fragment (data not shown). Taken at face value, the estimate of 11 generations (tb1 = ~11) is sufficient for the very smallest populations to have lost a substantial part of their genetic variation, and corresponds to 110–165 years. Accelerated forest clearance is known to have occurred in the sixteenth to nineteenth century, combined with a shift to livestock farming during the highland clearances (Dodgshon and Olsson 2006) and an increase in wild herbivores, partly as a result of the removal of wild predators (Holl and Smith 2007); these could plausibly have impacted dwarf birch numbers. This is also consistent with survey data showing substantial recent declines, presented in Fig. 1. One of populations, MU, produced an estimate of recent population size at the lower limit of its prior distribution. This disparity may be explained by its location: lowland and Southern populations are likely to have gone through earlier reductions in population size—which would be recovered in our model as a smaller Ne estimate.
Implications for dwarf birch conservation
Our results suggest that the long-term prospects for many dwarf birch populations in Scotland may be good, as there are levels of genetic diversity comparable to Scandinavia, despite fragmentation and reduced population size. Our ABC models suggest only moderate loss of diversity in the British populations upon post-glacial colonization, and that losses of diversity since then occurred in localized populations during their divergence from one another. Thus, different sub-sets of the ancestral variation of British B. nana populations are preserved in different fragments, so that overall levels of diversity in British B. nana populations are only moderately lower than the large continuous populations of Scandinavia. The long-term viability of B. nana in Britain may be enhanced by artificially restoring gene flow between populations, perhaps by culturing seedlings within, or transplanting among populations (Aitken and Whitlock 2013). Even very modest interventions may be effective.
The very smallest populations have diverged in allele frequency and lost genetic diversity at the genome-wide marker loci we surveyed. We can explain these effects as a consequence of recent severe isolation and reductions in effective population size. We would, therefore, expect loss of alleles conferring adaptive variation and inefficient selection to maintain traits at the local adaptive optimum. Consequently these populations are unlikely to harbor adaptive rear-edge diversity that needs to be conserved (sensu lato Hampe and Petit 2005). We, therefore, suggest that these populations could be enhanced by transplantation from the larger populations, but that it is unlikely to be beneficial to the larger populations to receive assisted gene flow from these small populations.
Broader utility of our approach
While, the potential for orders of magnitude more data from SNP analyses based on modern NGS technology is widely appreciated, comparative studies combining different categories of population genetic markers remain sparse (examples include Bradbury et al. 2015; Jeffries et al. 2016; Hodel et al. 2017). Here, we have found that for studies focusing on putatively neutral differentiation and drift, a modest number of SSR markers produce results that are comparable with thousands of SNPs (see also Hodel et al. 2016). We suggest that RAD-SSRs could be more widely used as they overcome many of the limitations of PCR-SSRs, yielding a high mutation-rate marker set, with >4 possible alleles per locus, and a well-specified ascertainment process that does not entail additional sequencing. In this way, we were able to exploit markers with different mutation rates to explore different demographic scenarios. Furthermore, comparing different types of markers can alert the researcher to biases introduced by the different ascertain processes (in our case possible effects on demographic estimates of contemporary population size and inbreeding), and evolutionary processes occurring at different rates. For conservationists where management interventions based on population data may influence the fate of species, robust conclusions based on multiple independent datasets are advisable.
Data archiving
-
Raw PCR-SSR data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.v75rj24.
-
Illumina read data from RADseq libraries has been uploaded to the European Nucleotide Archive project PRJEB26807, sample accessions ERS2598190- ERS2598376
-
Newly developed PCR-SSR primers are reported in the Supplementary Materials
-
RAD-SSR marker data mining script available on Github: https://github.com/JamesBorrell/RAD-SSR_extraction_scripts.git
-
Maximum Likelihood FST function available on Github: https://github.com/qmwugbt112/FstCalc
References
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44:367–388
Alsos IG, Eidesen PB, Ehrich D, Skrede I, Westergaard K, Jacobsen GH et al. (2007) Frequent long-distance plant colonization in the changing arctic. Science (80-) 316:1606–1609
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinforma 9:1–5
Aston D (1984) Betula nana L., a note on its status in the United Kingdom. Proc R Soc Edinb Sect B Biol Sci 85:43–47
Bacles CFE, Jump AS (2011) Taking a tree’s perspective on forest fragmentation genetics. Trends Plant Sci 16:13–18
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Balding DJ (2003) Likelihood-based inference for genetic correlation coefficients. Theor Popul Biol 63:221–230
Balding D, Nichols R (1995) A method for quantifying differentiation between populations atmulti-allelic loci and its implications for investigating identity and paternity. Genetica 96:3–12
Beaumont MA, Nichols RA (1996) Evaluating loci for use in the genetic analysis of population structure. Proc R Soc B Biol Sci 263:1619–1626
Bertorelle G, Benazzo A, Mona S (2010) ABC as a flexible framework to estimate demography over space and time: Some cons, many pros. Mol Ecol 19:2609–2625
Bradbury IR, Hamilton LC, Dempson B, Robertson MJ, Bourret V, Bernatchez L et al. (2015) Transatlantic secondary contact in Atlantic Salmon, comparing microsatellites, a single nucleotide polymorphism array and restriction-site associated DNA sequencing for the resolution of complex spatial structure. Mol Ecol 24:5130–5144
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: Building and genotyping loci de novo from short-read sequences. G3 Genes, Genomes, Genet 1:171–182
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Clausen KE (1966) Studies of compatibility in Betula. Jt Proc Second Genet Work Soc Am For Seventh Lake States For Tree Improv Conf. University of Minnesota, St Paul, MN pp. 48–52
Coates BS, Sumerford DV, Miller NJ, Kim KS, Sappington TW, Siegfried BD et al. (2009) Comparative performance of single nucleotide polymorphism and microsatellite markers for population genetic analysis. J Hered 100:556–564
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R et al. (2014) DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189
David P, Pujol B, Viard F, Castella V, Goudet J (2007) Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol 16:2474–2487
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Dodgshon RA, Olsson GA (2006) Heather moorland in the Scottish Highlands: The history of a cultural landscape, 1600-1880. J Hist Geogr 32:21–37
Doyle J, Doyle JL (1987) Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull 19:11–15
Eidesen PB, Alsos IG, Brochmann C (2015) Comparative analyses of plastid and AFLP data suggest different colonization history and asymmetric hybridization between Betula pubescens and B. nana. Mol Ecol 24:3993–4009
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: Implications for plant conservation. Annu Rev Ecol Evol Syst 24:217–242
Estoup A, Jarne P, Cornuet JM (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11:1591–1604
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Frankham R (1995) Effective population size/adult population size ratios in wildlife: A review. Genet Res (Camb) 89:491–503
Gilbert D, Di Cosmo L (2009) Towards restoration of treeline woodland and montane scrub. Bot J Scotl 55:177–187
Godwin H (1975) History of the natural forests of britain: Establishment, dominance and destruction. Proc R Soc B-Biological Sci 271:47–67
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 2:184–186
Gürcan K, Mehlenbacher SA, Botta R, Boccacci P (2010) Development, characterization, segregation, and mapping of microsatellite markers for European hazelnut (Corylus avellana L.) from enriched genomic libraries and usefulness in genetic diversity studies. TreeGenet Genomes 6:513–531
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: The rear edge matters. Ecol Lett 8:461–467
Hodel RGJ, Chen S, Payton AC, McDaniel SF, Soltis P, Soltis DE (2017) Adding loci improves phylogeographic resolution in red mangroves despite increased missing data: Comparing microsatellites and RAD-Seq and investigating loci filtering. Sci Rep 7:1–14
Hodel RGJ, Segovia-Salcedo MC, Landis JB, Crowl AA, Sun M, Liu X et al. (2016) The report of my death was an exaggeration: A review for researchers using microsatellites in the 21st century. Appl Plant Sci 4:1600025
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet 6:e1000862
Holl K, Smith M (2007) Scottish upland forests: History lessons for the future. Ecol Manag 249:45–53
Daniel C Ilut, Marie L Nydam, Matthew P Hare (2014) Defining Loci in Restriction-Based Reduced Representation Genomic Data from Nonmodel Species: Sources of Bias and Diagnostics for Optimal Clustering. BioMed Research International. 2014:1–9. https://doi.org/10.1155/2014/675158
Jeffries DL, Copp GH, Lawson Handley L, Olsén KH, Sayer CD, Hänfling B (2016) Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Mol Ecol 25:2997–3018
Jombart T (2008) Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Koskela J, Lefèvre F, Schueler S, Kraigher H, Olrik DC, Hubert J et al. (2013) Translating conservation genetics into management: Pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol Conserv 157:39–49
Kramer AT, Ison JL, Ashley MV, Howe HF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885
Kulju KKM, Pekkinen M, Varvio S (2004) Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). Mol Ecol Notes 4:471–473
Lampinen R, Lahti T (2016) Plant Atlas 2015. Helsinki University Museum of Natural History, Helsinki
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. NatMeth 9:357–359
Li Y-C, Korol AB, Fahima T, Beiles a, Nevo E (2002) Microsatellites: genomic distribution, putativa functions, and mutational mechanism: a review. Mol Ecol 11:253–256
Lischer HEL, Excoffier L (2012) PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Lowe AJ, Cavers S, Boshier D, Breed MF, Hollingsworth PM (2015) The resilience of forest fragmentation genetics[mdash]no longer a paradox[mdash]we were just looking in the wrong place. Hered (Edinb) 115:97–99
Martins K, Kimura RK, Francisconi AF, Gezan S, Kainer K, Christianini AV (2016) The role of very small fragments in conserving genetic diversity of a common tree in a hyper fragmented Brazilian Atlantic forest landscape. Conserv Genet 17:509–520
Martins WS, Lucas DCS, Neves KFDS, Bertioli DJ (2009) WebSat--a web software for microsatellite marker development. Bioinformation 3:282–283
Meirmans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24:3223–3231
Morin PA, Luikart G, Wayne RK (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216
Payseur BA, Cutter AD (2006) Integrating patterns of polymorphism at SNPs and STRs. Trends Genet 22:424–429
Piotti A (2009) The genetic consequences of habitat fragmentation: The case of forests. IForest 2:75–76
Priyam A, Woodcroft BJ, Rai V, Munagala A, Moghul I, Ter F et al. (2015) Sequenceserver: a modern graphical user interface for custom BLAST databases. bioRxiv: 033142.
R Development Core Team (2014) R: A language and environment for statistical computing. Found Stat Comput Vienna, Austria
Rousset F (2008) GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
RStudio Team (2015) RStudio: Integrated Development for R. RStudio, Inc, Boston, MA
Smout T, MacDonald AR, Watson F (2005) A history of the native woodlands of Scotland, 1500-1920. Edinburgh University Press, United Kingdom 2005, 434p
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Tipping R (1994) The form and fate of Scotland’s woodlands. Proc Soc Antiq Scotl 124:1–54
Truong C, Palme AE, Felber F, Naciri-Graven Y (2005) Isolation and characterization of microsatellite markers in the tetraploid birch, Betula pubescens ssp. tortuosa. Mol Ecol Notes 5:96–98
Tsuda Y, Nakao K, Ide Y, Tsumura Y (2015) The population demography of Betula maximowicziana, a cool-temperate tree species in Japan, in relation to the last glacial period: Its admixture-like genetic structure is the result of simple population splitting not admixing. Mol Ecol 24:1403–1418
Van Bers NEM, Van Oers K, Kerstens HHD, Dibbits BW, Crooijmans RPMA, Visser ME et al. (2010) Genome-wide SNP detection in the great tit Parus major using high throughput sequencing. Mol Ecol 19:89–99
Vallone PM, Butler JM (2004) AutoDimer: A screening tool for primer-dimer and hairpin structures. Biotechniques 37:226–231
Vranckx G, Jacquemyn H, Muys B, Honnay O (2012) Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol 26:228–237
Vucetich JA, Waite TA, Nunney L (1997) Fluctuating population size and the ratio of effective to census population size published by: society for the study of evolution stable. Source Evol 51:2017–2021. http://www.jstor.org/stable/2411022 URL:
Wang N, Borrell JS, Bodles WJa, Kuttapitiya A, Nichols Ra, Buggs RJa (2014) Molecular footprints of the Holocene retreat of dwarf birch in Britain. Mol Ecol 23:2771–2782
Wang N, Thomson M, Bodles WJa, Crawford RMM, Hunt HV, Featherstone AW et al. (2013) Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Mol Ecol 22:3098–3111
Wickham H (2009) ggplot2. Elegant graphics for data analysis. Springer: Springer-Verlag New York 210. https://doi.org/10.1007/978-0-387-98141-3
Wright S (1931) Evolution in mendelian populations. Genetics 16:97–159
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zhang Z, Gerstein M (2003) Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucl Acids Res 31:5338–5348
Zohren J, Wang N, Kardailsky I, Borrell JS, Joecker A, Nichols RA et al. (2016) Unidirectional diploid-tetraploid introgression among British birch trees with shifting ranges shown by restriction site-associated markers. Mol Ecol 25:2413–2426
Acknowledgements
This work was funded by NERC Fellowship NE/G01504X/1 to Richard Buggs. James Borrell was funded by NERC CASE studentship NE/J017388/1 in collaboration with Trees for Life and Highland Birchwoods. Nian Wang was funded by the Chinese Scholarship Council. Fieldwork in Scandinavia was funded by an EU-INTERACT Transnational Access grant. We gratefully acknowledge the assistance provided by Dr. Billy Bodles, Alan Watson Featherstone, and members of the Montane Scrub Action Group and numerous landowners who permitted access to their estates.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borrell, J.S., Wang, N., Nichols, R.A. et al. Genetic diversity maintained among fragmented populations of a tree undergoing range contraction. Heredity 121, 304–318 (2018). https://doi.org/10.1038/s41437-018-0132-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0132-8
This article is cited by
-
Using genome-wide diversity and population structure to define management units in the cirio (Fouquieria columnaris), an emblematic tree of the Sonoran Desert
Biodiversity and Conservation (2023)
-
Population genomics study for the conservation management of the endangered shrub Abeliophyllum distichum
Conservation Genetics (2022)
-
Fine-scale analysis reveals a potential influence of forest management on the spatial genetic structure of Eremanthus erythropappus
Journal of Forestry Research (2021)
