
Abstract
Admixture resulting from natural dispersal processes can potentially generate novel phenotypic variation that may facilitate persistence in changing environments or result in the loss of population-specific adaptations. Yet, under the US Endangered Species Act, policy is limited for management of individuals whose ancestry includes a protected taxon; therefore, they are generally not protected under the Act. This issue is exemplified by the recently re-established grey wolves of the Pacific Northwest states of Washington and Oregon, USA. This population was likely founded by two phenotypically and genetically distinct wolf ecotypes: Northern Rocky Mountain (NRM) forest and coastal rainforest. The latter is considered potentially threatened in southeast Alaska and thus the source of migrants may affect plans for their protection. To assess the genetic source of the re-established population, we sequenced a ~ 300 bp portion of the mitochondrial control region and ~ 5 Mbp of the nuclear genome. Genetic analysis revealed that the Washington wolves share ancestry with both wolf ecotypes, whereas the Oregon population shares ancestry with NRM forest wolves only. Using ecological niche modelling, we found that the Pacific Northwest states contain environments suitable for each ecotype, with wolf packs established in both environmental types. Continued migration from coastal rainforest and NRM forest source populations may increase the genetic diversity of the Pacific Northwest population. However, this admixed population challenges traditional management regimes given that admixture occurs between an adaptively distinct ecotype and a more abundant reintroduced interior form. Our results emphasize the need for a more precise US policy to address the general problem of admixture in the management of endangered species, subspecies, and distinct population segments.
Similar content being viewed by others

Introduction
A complication for the conservation of rare and endangered species is the level of protection for admixed populations. Current policy and management protocols favour the biological species concept, where speciation is defined as descent with modification in a reproductively isolated lineage (Dobzhansky 1935; Mayr 1947), and lack guidelines with regards to admixed populations (reviewed in Jackiw et al. 2015). Consequently, adequate protection may not be granted to taxa that experience a high frequency of gene flow and introgression over the course of their evolutionary histories (Rhymer and Simberloff 1996). Yet, gene flow across species and populations is a natural phenomenon that happens often and may be critical for evolutionary processes (reviewed in Slatkin 1987; Twyford and Ennos 2012; Abbott et al. 2013). Natural admixture allows for the preservation of the historical genetic connectivity between populations. Although admixture may result in the loss of population specific adaptations (e.g. Rhymer and Simberloff 1996; Muhlfeld et al. 2009), it may also provide the genetic variation on which selection can act (e.g., Smith et al. 1997b; Hedrick 2013). If admixture provides genetic variation, it may be as important to maintaining adaptive potential, the capacity for future evolutionary change, as evolution by natural selection or genetic drift within reproductively isolated lineages (Arnold 2016; vonHoldt et al. 2017). This concept of species as fixed entities with complete reproductive isolation is currently used in many cases of species management. However, the “web-of-life” (WOL) framework is a more realistic concept that acknowledges that horizontal gene transfer through hybridization, introgression and reticulate evolution is prevalent in some systems (Arnold and Fogarty 2009). Therefore, this framework includes adaptive potential by protecting genomic and phenotypic diversity, ecological function and resilience and does not preclude an individual, population or species from protection due to natural admixture (Arnold 2016; vonHoldt et al. 2017).
Appropriate management and protection of natural re-colonized and admixed populations involving an endangered source is a critical challenge that requires case-by-case solutions (Allendorf et al. 2001; vonHoldt et al. 2017). Each context requires the initial detection and identification of hybrid individuals, which is not a trivial task (Allendorf et al. 2001). Recent advances in high-throughput genomic resources have greatly increased our ability to detect and characterize admixture in hybrid populations (Allendorf et al. 2010; Hohenlohe et al. 2011; vonHoldt et al. 2013; Abbott et al. 2016; Wayne and Shaffer 2016) and provide evidence of natural movement of genetic variation across populations and species (the WOL framework; vonHoldt et al. 2017). Once the occurrence of natural admixture is established, two questions should be answered before management decisions are made, as suggested by Wayne and Shaffer (2016). First, do admixed individuals perform ecosystem functions and services that are similar to those performed by the endangered entity? Second, would habitat restoration for the native endangered entity enable natural selection to increase the proportion of genetic variants characteristic of the native endangered entity (Wayne and Shaffer 2016)? These questions as well as the WOL framework establish an inclusive foundation for the management of admixed populations in the light of evolutionary and ecological principles.
One case of natural admixture that may highlight the importance of the WOL framework is that of the North American grey wolf (Canis lupus). The natural re-colonization of previously extirpated populations of large carnivores is rare but has been documented in wolves in North America and Europe (Chapron et al. 2014). Historically, wolves were common in the Pacific Northwest (PNW) region of North America (Bailey 1936; Young and Goldman 1944) but were extirpated in the US portion by the mid-1930s (Bailey 1936; Verts and Carraway 1998). Wolves naturally re-colonized northwest Montana (MT), USA, from Alberta (AB) and British Columbia (BC), Canada, in the mid-1980s (Boyd et al. 1995). More recently, wolves naturally re-colonized the states of Oregon (OR) and Washington (WA), USA, and as of 2015, 13 and 18 packs inhabit those states, respectively. Given that individuals disperse an average of 50–100 km or up to several hundred kilometres before establishing territories (Mech 1970; Fritts 1983; Merrill and Mech 2000; Jimenez et al. 2017), these re-established wolves in OR and WA are suspected to be migrants from adjacent wolf populations that consist of two ecotypes, the coastal (referred to as rainforest wolves in some previous studies) and the Northern Rocky Mountain forest (NRM) populations. These two ecotypes represent locally adapted and specialized wolves with respect to den-site use, foraging habits, physiology and prey specialization (Fritts et al. 1995; Mladenoff et al. 1995; Paquet et al. 1996; Mladenoff et al. 1997; Mladenoff and Sickley 1998; Haight et al. 1998; Mladenoff et al. 1999; Callaghan 2002) and exhibit environmentally driven genetic differences between coastal and NRM populations (Weckworth et al. 2005; Muñoz Fuentes et al. 2009; Weckworth et al. 2011; Schweizer et al. 2016a, b). Previous studies aimed at identifying ecotype-specific selection in NRM and coastal wolves found multiple signals of selection on genes related to dentition, diet, metabolism, musculature, organismal system, skeletal morphology and vision (Schweizer et al. 2016a, b). Furthermore, the coastal wolf population often has high allelic differentiation from all other populations, especially for candidate single-nucleotide polymorphisms (SNPs) under selection (Schweizer et al. 2016a, b).
The origins of some of the WA and OR population are from geographically proximate NRM wolves that in turn derive from wolves reintroduced to Idaho (ID) from northern, interior BC and AB as well as the naturally re-established wolves from MT (Jimenez et al. 2017). However, it is unknown if immigration, territory establishment and subsequent breeding of coastal wolves in the PNW has also occurred. At least two important questions currently remain unresolved: (1) is genetic admixture between divergent lineages (ecotypes) occurring in the re-established PNW population?; and (2) can the habitat of PNW region support these two ecotypes in a potential admixture zone?
Any potential admixture may have conservation implications for the wolves of the PNW region. Coastal wolves comprise genetically contiguous populations in coastal BC and the Alexander Archipelago in southeast Alaska (AK, USA; Weckworth et al. 2010, 2011). The Alexander Archipelago wolves were considered for protection under the USA Endangered Species Act (ESA) due to human-mediated habitat alteration that resulted in a 60% decline in the population over 1 year (Toppenberg et al. 2015). Although this wolf population was ultimately not listed, it is still impacted by legal and illegal hunting and deserves special consideration as a unique ecotype not found outside this area (Muñoz Fuentes et al. 2009; Schweizer et al. 2016a, b). Furthermore, admixture is likely recent and therefore not yet in equilibrium, thus warranting protection as the populations stabilize. If Alexander Archipelago wolves attain protected status under ESA, then admixture, if found, should influence how the wolves of the PNW are managed. Protection status should be granted given several conditions: (a) admixture is a result of natural patterns of wolf dispersal; (b) historical genetic connectivity is preserved (Hendricks et al. 2015); and (c) adaptive potential is maintained, following the WOL framework (Wayne and Shaffer 2016; vonHoldt et al. 2017).
We used three complementary approaches to assess the source populations of PNW wolves and their suitability to areas of reintroduction. First, we sequenced a portion of the mtDNA control region in wolves from WA, OR, and surrounding populations to establish maternal lineages. Second, we used SNPs obtained through targeted DNA capture and sequencing to estimate local population structure, ancestry and relatedness among individuals. Third, we assessed habitat preference of re-established wolf packs in the PNW region. To do so, we used ecological niche models (ENMs) based on climate predictors to identify appropriate habitat for the NRM and coastal wolf ecotypes and then mapped centroid locations of existing WA and OR packs (as of 2015) to assess potential genetic barriers associated with environmental differences. Results from these approaches establish the source populations for naturally re-established wolf populations and better inform the conservation and management of the wolf populations in WA and OR.
Materials and methods
Sample collection
Tissue and/or blood samples from grey wolves were collected in WA by the Washington Department of Fish and Wildlife (WDFW) and in OR by the Oregon Department of Fish and Wildlife (ODFW) during radio collaring efforts of live animals or from carcasses. We analysed 32 samples from OR wolves (collected by ODFW 2009–2013) and 22 samples from WA (collected by WDFW 2008–2012). Scat or bone and tissue samples from wolf carcasses in coastal BC were collected as a part of a long-term study (Darimont et al. 2008). Appropriate permits were granted to researchers for sample collection. Additional samples from interior Yellowstone National Park (YNP), ID, MT, BC, AB, and coastal BC and Alaska were selected from a set of samples of grey wolves used in previous studies (vonHoldt et al. 2010, 2011). DNA was extracted from tissue, blood, bone and scat samples using the standard commercial kit protocols (Qiagen DNA QiaAmp minikit and EZNA Stool Kit from Omega Biotek).
Species determination
To confirm the species and subspecies status of samples, a panel of 24 species-diagnostic markers was used to resolve the contributions of ancestry from the grey wolf (C. lupus), domestic dog (C. lupus familiaris) and coyote (C. latrans) and resolve first- and second-generation dog–grey wolf hybrids (vonHoldt et al. 2013). These markers (20 resolving wolf vs. dog, two resolving dog vs. coyote and two resolving coyote vs. wolf) were identified and confirmed against a panel of 832 dogs, 180 grey wolves and 53 coyotes analysed on the Affymetrix Canine SNP v2. microarray (vonHoldt et al. 2013). The markers were assayed using a quantitative polymerase chain reaction (qPCR) High-Resolution Melting (HRM) assay and Roche LightCycler 480 instrument (Indianapolis, IN). Two dog, two western coyote and two western Canadian grey wolf samples were used as non-admixed references for allele calls. The qPCR reaction mixes and thermocycler conditions followed vonHoldt et al. (2013). HRM melt temperatures were analysed using the Roche LightCycler 480 Software v1.5.0.
Mitochondrial haplotype determination
To further confirm the species or subspecies status of samples, eliminate possible coyote/wolf or dog/wolf hybrid individuals and assess the distribution of mtDNA haplotypes, a 318-bp portion of mitochondrial control region (MT-CR) was amplified using two sets of overlapping primer pairs: (i) Thr-L (Vilà et al. 1999) and ddl5R (Leonard et al. 2002); and (ii) ddl1F and ddl2R following the protocol in Leonard et al. (2002). Amplified PCR products were sequenced in both directions using BigDye on an ABI3730XL capillary sequencer (Applied Biosystems, Inc). Sequences were visualized and aligned in Geneious 6.0.5 (Biomatters). A local BLAST search (Altschul et al. 1997) was performed on individual consensus sequences against Genbank partial MT-CR haplotypes (n = 75 globally distributed grey wolves; n = 125 coyotes; n = 1 red wolf (C. rufus); n = 30 domestic dogs). Putative assigned haplotype matches were confirmed by BLAST searches against the Genbank reference nucleotide database.
Capture array library preparation
To determine the source populations of individuals from PNW, 96 individuals (coastal BC—8; inland BC—5; MT—23; ID—17; YNP—6; WA—16; OR—21) were sequenced using a custom capture array designed to target sequences from 1040 candidate genes and 5073 1 kb neutral regions from the dog reference genome (CanFam3.1; Schweizer et al. 2016a). Putatively neutral regions were identified using methods described by Freedman et al. (2014) and for which there exists a precedent in humans (Wall et al. 2008) and wolves (Schweizer et al. 2016a). First, genic regions from the dog reference genome (CanFam3.1) were identified using annotations from the union of refGene, Ensembl and SeqGene annotation databases. All annotated transcripts had proper start and stop codons and contained no internal stop codons. Second, 1 kb neutral regions were chosen using the following characteristics: (1) minimally 100 kb from any known or predicted genes (based on observed levels of linkage disequilibrium (LD) in wolves (Gray et al. 2009); (2) not located within highly repetitive regions of the dog genome; (3) uniquely mapping regions of the genome as computed by TALLYMER (Kurtz et al. 2008); (4) phastCons scores <0.5 (Siepel et al. 2005); and (5) GC content within two standard deviations of the mean dog genome GC content.
DNA quantity and quality were assessed with the Qubit Fluorometer High Sensitivity Kit and visualization after electrophoresis on a 2% agarose gel, respectively. Samples with at least 600–1000 ng of dsDNA and a molecular weight of >1 kb were sheared to ~ 300–500 bp using a Bioruptor NGS Sonication System (Diagenode). Sequencing libraries for each individual were prepared using a with-bead library preparation protocol (Faircloth et al. 2013) that included labelling with a unique 6-bp index (Faircloth and Glenn 2012). Two individual libraries were pooled and allowed to hybridize to the array for 24 h. Each pool was target-enriched and PCR-amplified according to the MYbaits protocol (MYcroarray), with modifications as in Schweizer et al. (2016a). Before sequencing, 24 individuals (12 capture libraries) were pooled and enriched libraries were run on two lanes with 100-bp paired-end sequencing on an Illumina HiSeq 2000.
Sequence alignment and processing
The Broad Institute GATK v2.6-4 “Best Practices” pipeline (https://software.broadinstitute.org/gatk/best-practices/) was used for sequence alignment and processing. Demultiplexed fastq reads that passed the Illumina filter using fastq_illumina_filter 0.1 (http://cancan.cshl.edu/labmembers/gordon/fastq_illumina_filter/) were trimmed for adapter sequences and a minimum base quality of 20 using trim_galore 0.3.1 (http://bioinformatics.babraham.ac.uk/projects/trim_galore/). Aligned forward and reverse reads were mapped to the reference dog genome (CanFam3.1) using bwa aln (seed length of 28) and bwa sampe (insert size of 1000 bp; (Li and Durbin 2009). Once duplicates were removed using samtools rmdup, a local realignment was completed using GATK 2.6-4 (DePristo et al. 2011). Mate information was fixed with picard tools (http://picard.sourceforge.net). GATK Base Quality Score Recalibration (BQSR) was performed using the –knownSites flag with the final SNP set from Schweizer et al. (2016a). After adding in 38 samples from coastal BC, coastal AK, AB and YNP that were previously enriched and sequenced using the same protocols (see Supplementary Table S1; Schweizer et al. 2016a, 2018), SNPs were called using the GATK Haplotype Genotyper algorithm.
Array variant filtering and final sample set
Variant filtration was completed using ten filter expressions recommended by the GATK “Best Practices” pipeline. Variants with a depth of coverage >10 and minimum genotype quality >30 were kept in the final data set. The VCFtools package (Danecek et al. 2011) was used to assess the quality of filtered and aligned reads. Sites called in <95% of individuals were subsequently removed from further analysis. The number of segregating sites and mean coverage per individual was calculated using VCFtools. Data sets were LD-pruned using PLINK (-indep-pairwise 50 5 0.5; Purcell et al. 2007). We used the programs KING v1.4 (Manichaikul et al. 2010) and PRIMUS v0.5 (Staples et al. 2013) to calculate relatedness and then removed one individual per related pair with a pairwise identity-by-state greater than or equal to 0.5, calculated from the LD-pruned data set. We used four data sets for subsequent analyses: (1) all individuals, all loci; (2) all individuals, putatively neutral loci; (3) unrelated individuals, all loci; and (4) unrelated individuals, putatively neutral loci.
Population structure, individual assignment and gene flow estimates
To verify genetic differentiation between ecotypes, we used VCFtools to calculate Weir and Cockerham’s (1984) θ, an estimator of FST (Wright 1951). To genetically assign and determine ancestry of WA and OR wolves to YNP, ID, MT, AB, interior BC or coastal BC populations, we applied two clustering methods to both SNP data sets with unrelated individuals. First, principal component analysis (PCA) was performed using SMARTPCA within EIGENSTRAT v3.0 (Price et al. 2006). Second, using the default settings, we applied the program ADMIXTURE (Alexander et al. 2009) to partition and classify individuals into K = 1 through K = 10 clusters. Inbreeding coefficients (FIS) were also calculated using VCFtools.
We used two methods to identify potential related individuals across state boundaries and estimate levels of gene flow. We used the data set with all individuals and putatively neutral, LD-pruned loci. First, pairwise relatedness values >0.1 were used to identify individuals that may have shared ancestry with individuals from other geographic locations sampled in this study. Second, we used the program BayesAss v3.0.4 (Wilson and Rannala 2003), which is a Bayesian assignment test that estimates individual ancestry. The program was run for 10 million Markov chain Monte Carlo (MCMC) iterations with the first 1 million iteration discarded as burnin and sampling every 100 iterations. Adjusted mixing parameters for migration rate (m = 0.2), allele frequency (a = 0.5) and inbreeding coefficients (f = 0.002) were used to optimize the acceptance rate.
Ecological niche modelling
Preparation of ecotype occurrence data
Given the genetic differentiation between ecotypes and adaptive distinction of the coastal wolves (Muñoz Fuentes et al. 2009; Schweizer et al. 2016a, b), we predicted the optimal environmental niches of the coastal and NRM forest ecotypes to assess whether environmental differences occur within the PNW region and whether habitat differences between coastal and NRM forest act as barriers to gene flow for the re-established packs in WA and OR. Non-duplicate localities for coastal wolves (coastal BC and southeast Alaska; n = 20) and NRM forest wolves (interior BC, AB, MT, WY, ID; n = 119) were compiled using data from the Global Biodiversity Information Facility portal (www.gbif.org) and voucher museum specimens (Supplementary Figures S7 & S8). These localities do not include occurrence points of individuals sampled for DNA. When exact geographic coordinates for a specimen were not available, we used the provided location name to estimate the geo-reference for the individual. Localities for which geo-referencing could not be defined more precisely than the level of county or similar administrative unit were excluded.
Preparation of environmental data
Bioclimatic variables from WorldClim v1.4 (Hijmans et al. 2005) were selected according to their roles in determining the physiological limits of species (e.g., variation in annual means, extremes and seasonality of temperature and precipitation). These bioclimatic variables (n = 19) are at a 1 km resolution and metrics are derived from monthly interpolated temperature and rainfall climatologies spanning the years 1950 to 2000 (Hijmans et al. 2005). For each wolf ecotype, clusters of highly correlated variables were identified and removed to trim variables that were not contributing to the model (Harrigan et al. 2014). The nine variables used in both models were: annual mean temperature, annual precipitation, precipitation seasonality, precipitation of the warmest quarter, precipitation of the coldest quarter, mean diurnal range, temperature seasonality, maximum temperature of the warmest quarter, and maximum temperature of the coldest quarter. Elevation (SRTM) was also used in both models. Current vegetation data were not included in this analysis because vegetation patterns are more intensively influenced by anthropogenic activities (e.g. deforestation, land cover conversion, urban development and road network intensification) and therefore could change quickly. Additionally, because samples spanned multiple years, accurate vegetation data could not be obtained.
MaxEnt modelling
We ran MaxEnt v3.3.3k (Phillips et al. 2006), which uses a probabilistic framework, to model the environmental niches from occurrence data (described above). Its main assumption is that the incomplete empirical probability distribution (which is based on the species occurrences) can be approximated by a probability distribution of maximum entropy (the MaxEnt distribution) subject to certain environmental constraints and that this distribution approximates a taxon’s potential geographic distribution (Phillips et al. 2006). The use of MaxEnt is advantageous in the study of endangered taxa, for which locality data may be sparse, as it performs well with only a small number of point localities (Jordan and Ng 2002; Hernandez et al. 2006; Wisz et al. 2008). Unlike many other algorithms, it requires only presence data to assign spatially explicit probabilities of occurrence (Phillips et al. 2006) and it consistently ranks high in inter-model comparisons (Elith et al. 2006; Diniz-Filho et al. 2009; Harrigan et al. 2014). Further, several recent studies have shown that MaxEnt performs successfully in modelling the distribution of motile species (Rodríguez Soto et al. 2011; Lv et al. 2011; Blair et al. 2013).
In this study, we used the MaxEnt default settings of convergence threshold (10−5) and 100 cross-validated replicates. This cross-validation replicate process involved the random splitting of occurrence data into a number of equal-sized groups, known as “folds”, where models were created leaving out one fold for each run. For each replicate, the excluded fold is used to evaluate the model (Phillips et al. 2006). The study area over which the potential distribution is computed, and from which the MaxEnt algorithm samples “background” points to train the model, are substantially larger than the known ranges of the ecotypes. We verified that modelling results were insensitive to the choice of study area size by building models with progressively larger study areas, increased at an increment of 5° latitude and longitude (data not shown). Regularization attempts to balance model fit and complexity, with the default setting multiplying each automatic regularization parameter by 1. Additional multiplication of these parameters tends to smooth (make the model more generalized) at the expense of model fit (Elith et al. 2011). For comparisons of models, we chose to leave regularization parameters the same across all runs (r = 1), particularly because default settings represent a conservative approach to estimating species distributions based on occurrences. Using ENMeval (Muscarella et al. 2014), we tested for the effects of spatial autocorrelation and model complexity by running each ecotype occurrence data set through several different partitions as recommended by the authors.
MaxEnt produces a continuous prediction with values ranging from 0 to 1 (in units of probability of occurrence) indicating least suitable to most suitable conditions for the taxa under consideration (Phillips et al. 2006). To convert this continuous output into a binary prediction that approximates the potential distribution, we used a probability threshold equivalent to the minimum predicted probability of occurrence at actual occurrence localities used to train the model (Phillips et al. 2006).
Testing model performance
Model performance was evaluated by the area under the curve (AUC), which is often used to measure model performance (Rödder et al. 2009; Harrigan et al. 2010; Fourcade et al. 2014; Sesink Clee et al. 2015). AUC values were calculated by comparing model performance to a random model of associations between presence localities and environmental predicting factors (DeLong et al. 1988). AUC values range from 0.5 to 1.0; with values close to 0.5 corresponding to a model that is no better at predicting an ecological niche than a random model and a value of 1.0 corresponding to a model with a perfect fit.
Probability of occurrence
As of 2015, 31 wolf packs inhabited the PNW states of WA (n = 18) and OR (n = 13). The centroid location of each pack was used as the proxy for area used by each pack. The resulting ENMs were used to calculate the probability of each PNW wolf pack’s occurrence in coastal or interior environments.
Results
Species determination and mitochondrial haplotype determination
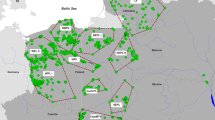
Genotypes from the panel of 24 species-diagnostic markers identified all modern samples as pure grey wolf, with no evidence of recent dog or coyote ancestry. Therefore, we sequenced 139 wolf samples for a 318 bp fragment of the mitochondrial control region to determine haplotype distributions (Fig. 1, Supplementary Table S1 and S2). Relative to other North American populations, the diversity of maternal lineages in PNW wolves was slightly lower than average (H = 4; Fig. 1 & Supplementary Table S2, average in NA = 4.5; Table 5 in Chambers et al. 2012). We have not included the Great Lakes or eastern Ontario populations in calculations due to taxonomical conflicts and admixture events with coyotes. Interestingly, two WA individuals had haplotype lu68 (Fig. 1 and Supplementary Tables S1 and S2), which is otherwise known only from populations in coastal BC (current study: N = 4 of 29; Fig. 1 and Supplementary Tables S1 and S2) and previous studies (Muñoz Fuentes et al. 2009, 2010; Weckworth et al. 2010). Consequently, the mitochondrial haplotype analysis confirms our diagnosis that the samples are of grey wolf ancestry and suggests some gene flow from the coastal ecotype (coastal BC and southeast AK) to the population in WA with the principal influence from NRM wolves.

Capture array sequences
We obtained high-quality sequence reads with a per-individual average unfiltered yield of 2254.62 ± 954.12 Mb, 92.92 ± 3.74% raw reads passing Illumina filters and a mean quality of 36.90 ± 1.84. After processing and removing low-quality reads, 80 ± 16.3% of raw reads mapped uniquely to the dog reference genome (i.e. after PCR duplicate removal). After genotyping, quality filtering and removing low coverage (<10×) individuals (n = 9), the mean depth of coverage over all regions on the capture array was 89.79 ± 35.13 (Supplementary Figure S1), with a mean depth of coverage over neutral regions of 137.95 ± 52.33 (Supplementary Figure S1). Mean depth of coverage for scat samples from coastal BC over all regions was 22.37 ± 26.94 and ranged from 0.99 to 59.89 (Supplementary Figure S2), with a mean neutral depth of coverage of 34.23 ± 40.87 that ranged from 1.52 to 92.64 (Supplementary Figure S2). We excluded data from half (n = 3) of our original faecal samples due to low coverage (<10×) that may be a result of complications with DNA extraction and library preparation (see Discussion in Supplementary File 1).
After filtering genotypes, we separated data into two sets consisting of all variant loci (92,296 SNPs) and variant loci within neutral regions (41,735 SNPs). The transition to transversion ratio for all regions was 2.31 and for neutral regions was 2.23, which is similar to previously reported values in wolves (Freedman et al. 2014; Zhang et al. 2014; Schweizer et al. 2016a). After LD-pruning, there were 35,406 variable positions, of which 18,508 were within neutral regions. After removal of one individual per related pair (parent–offspring or full siblings; N = 43 individuals), the remaining set of 83 individuals included 9 ID, 13 MT, 28 YNP, 7 AB, 9 coastal BC, 4 interior BC, 5 WA and 8 OR wolves.
Population structure, individual assignment and gene flow estimates
We calculated pairwise FST between population pairs to verify that that there is differentiation between ecotypes using our data set. Mean among-population pairwise FST of the 18,508 LD-pruned neutral SNPs was moderate. The coastal wolves were the most differentiated by this measurement with pairwise FST values ranging from 0.104 between coastal and WA to 0.170 between coastal and MT populations (Table 1). MT was the second most differentiated population with FST values that ranged from 0.034 (YNP) to 0.072 (AB). Excluding coastal wolves, FST values ranged from 0.001 between interior BC and WA to 0.072 (between AB and MT, Table 1). Inbreeding coefficients (FIS) were near zero for all populations (FIS = −0.0115 to 0.0425; Supplementary Table S3), except the coastal BC population (FIS = 0.1116; Supplementary Table S3).
To assess the genetic partitions based on SNP data, we used complementary analyses of genetic clustering patterns. Plots of the first two components from PCA show several distinct clusters that correspond with sampling location (Fig. 2). On both PC1 and PC2, the individuals of the coastal BC and AK population cluster distinctly, with no other samples occurring within the 95% confidence interval (CI) (Fig. 2). The NRM populations (BC, AB, ID, YNP, MT and OR) are more continuous in PC space with less distinct clustering than the coastal population. The OR samples cluster together and are spatially between the YNP and MT clusters. MT individuals form a 95% CI cluster that includes one known migrant from ID (vonHoldt et al. 2010), one individual from interior BC and most of the WA individuals. Three WA individuals fall intermediate to the coastal population and the NRM populations. PCA results were similar between all loci and neutral loci for both data sets (all 126 and 83 unrelated individuals; Fig. 2; Supplementary Figure S3).

a PCA plot of all 126 wolves for LD-pruned data set (18,508 SNPs) with 95% confidence intervals. b PCA plot of 83 unrelated wolves for same LD-pruned data set with 95% confidence intervals. Population abbreviations as follows: AB Alberta, Canada; BC interior British Columbia, Canada; cBC coastal British Columbia, Canada and Alaska, USA; ID Idaho, USA; MT Montana, USA; OR Oregon, USA; WA Washington, USA; YNP Yellowstone National Park, USA
Our second approach to assess genetic clustering used ADMIXTURE, which showed the best-supported number of clusters equal to 3 as evident by the lowest cross-validation error rate (Fig. 3, Supplementary Figures S4, S5). These three clusters represent the naturally re-established MT population, the reintroduced YNP and ID population and the coastal population (Supplementary Figure S5). However, the AB individuals form an additional, biologically relevant cluster at K = 4 (Supplementary Table S4). Therefore, we present assignment proportions from the results of K = 4. Two WA individuals have high assignment (>49%) to the AB cluster, with signatures of ancestry to the coastal population. The other three WA individuals assign to the MT cluster (>90%). All OR individuals have ancestry from NRM populations. Of the four YNP individuals that strongly assign to the AB cluster, three are founders (assign to the AB cluster with >67% ancestry) and were reintroduced from northern AB and BC populations (vonHoldt et al. 2010). Four ID individuals assign to the AB cluster (>50%), three of which are founders and assign to the AB cluster with >88.5% ancestry. Given our data set included samples spanning multiple time points occurring during wolf introduction, we wanted to test for temporal effects on the number of genetic clusters. ADMIXTURE analysis was re-run under the same parameters after removing known, unrelated founders from YNP (n = 3) and ID (n = 3). The same four clusters are observed after removing the founders (Fig. 3), which indicates that there is little detectable temporal effect on allele frequencies per population. The cross-validation error values for ADMIXTURE runs of unrelated individuals for all data or only neutral data indicate an optimal K = 3, as described above (Supplementary Figure S4). When the coastal individuals were excluded from ADMIXTURE analysis (Supplementary Figure S6), K = 2 had the lowest CV error for both sequence data sets. This result further suggests that the coastal ecotype is a valid cluster.

Population assignment at K = 2 to K = 5 for 75 unrelated individuals (after removing founder individuals from YNP and ID), as determined by running admixture on a set of 18,508 LD-pruned SNPs within neutral regions. The lowest cross-validation error rate occurred at K = 3, which shows the naturally re-established MT population, the reintroduced YNP and ID population and the coastal population. Higher values of K are also biologically meaningful and therefore shown
To identify individuals that might be related across state boundaries, we filtered the pairwise relatedness data to include pairs related above 0.1 and from different populations (Supplementary Table S5). Individuals from AB and coastal populations (including coastal BC and AK wolves) did not have pairs from differing populations with relatedness values above 0.1. All other populations were included in this filtered subset. At the relatedness level of ~0.5 (full sibling or parent–offspring), one WA individual (WA010817_WA) was related to three OR individuals (OR11_OR, OR16_OR, OR10_OR). Individuals from MT, ID and YNP were found to have levels of relatedness from 0.1 to 0.5. This finding supports previous work highlighting gene flow among wolf populations of the Northern Rocky Mountains (vonHoldt et al. 2010). Interestingly, one OR wolf (OR1_OR) has relatedness of 0.13 and 0.17 with two YNP founders (033F and 040F, respectively). These two YNP founders are themselves unrelated and from different packs, but owing to limited sampling of additional related YNP wolves, the exact ancestry of OR1_OR cannot be determined.
Results for the Bayesian estimation for non-symmetrical rates of gene flow found the proportion of individuals per generation originating from within each identified cluster varied from 82.19 to 92.36%, with the highest value found in the MT cluster (Supplementary Table S6). Each independent run of BayesAss converged towards similar values of logProb despite different starting seeds. Moreover, visualization of the MCMC trace output confirmed convergence and the posterior probability values of migration suggests strong isolation for all the inferred clusters (not shown). BayesAss estimated that 16.93% of individuals in interior AB and BC migrated from the reintroduced YNP/ID population (Supplementary Table S6). MT contributes 6.22%, 6.67% and 5.50% migrants to OR, WA and BC/AB clusters, respectively. OR received 7.13% migrants from the YNP/ID (reintroduced) cluster. WA received 5.02% migrants from the coastal cluster. There is little migration among other clusters (<2% of the population migrating per generation; Supplementary Table S6).
Ecological niche modelling
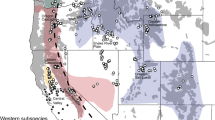
Aggregate ENMs were produced by averaging values from 100 replicate iterations of the data for the coastal and NRM forest wolf ecotypes niche models (Supplementary Figures S7–S8, respectively). Training and test AUC values for both models were as high as 0.99 (model AUCs ranged from 0.75 to 0.99, depending on partition scheme used, see Supplementary Figures S9, S10), which suggests that the models were highly informative and describe climatically suitable areas that correspond well with the environmental conditions of localities with known ecotypes. The composite model revealed complete optimal environmental niche divergence for each of these ecotypes with very little geographic overlap between ecotypes and concordance in model probability of occurrence regardless of data partitioning scheme (Fig. 4, Figures S9 and S10). Suitable habitat analysis would additionally consider prey and human population densities, percentage of forest cover and forest composition, as well as interference from roads and urbanized land cover, which is out of the scope of the current study.

Composite MaxEnt distribution model for coastal and interior wolves within the area of the natural re-colonization and potential admixture zone. Warmer colours correspond to the most suitable environment for interior wolves and cooler colours correspond to most suitable environment for coastal wolves. As of 2015, 31 wolf packs inhabited the PNW states of Washington (n = 18) and Oregon (n = 13). Centroid location and pack name of these packs are plotted to show re-colonization of these states but were not used to inform the models. Wolves have been observed in the more coastal areas on the western side of WA but have not established packs as of the end of 2017 (https://wdfw.wa.gov/conservation/gray_wolf/reporting/sightings.html). Full MaxEnt distribution models for coastal and interior wolves are available in Figures S7 and S8
Using centroid pack locations and the aggregate ENMs, the likelihood that a pack occurs in the coastal environment or the interior environment was calculated (referred to as probability of presence throughout; see Elith et al. 2011). Of the 18 WA wolf packs, 17 packs have a greater probability of presence in interior environment than in coastal environment indicating more association of wolves with the interior environment based on our models (Supplementary Table S7; Fig. 4). However, the Teanaway pack, the most western pack currently in WA, has a greater probability of presence in the coastal habitat than the interior habitat (Fig. 4). No DNA samples were obtained from the Teanaway Pack and we do not currently know the genetic ancestry of this pack. The Lookout pack in WA was on the boundary of interior and coastal habitat and contained a wolf with mtDNA evidence for ancestry to the coastal population and admixed nuclear ancestry of 45% AB and 49% coastal wolf (Sample: RKW4318; Supplementary Table S4). The Wedge pack has a greater probability of presence in the interior habitat (Fig. 4), yet contained an individual (Sample: WAWedge8) with coastal mtDNA ancestry and admixed nuclear ancestry of 53% AB, 35% coastal and 11% MT (Supplementary Table S4). Of the 13 OR wolf packs, all but one, the Rogue pack, have a higher interior probability of presence than coastal habitat. The Rogue pack has a very low (0.0247–0.0476) probability of presence in both habitats with a slightly higher probability of presence in coastal habitat (Fig. 4). Data from GPS-radio collar tracking devise indicate that this pack was established from a male disperser from the Imnaha pack (NE Oregon) and mated with a female likely from the Snake River or Minam packs (NE Oregon). Unfortunately, we were not able to obtain DNA samples for genetic ancestry analysis of any individuals from the Rogue pack.
Discussion
Our results confirm prior work on population structuring of wolves in western North America (Carmichael et al. 2007; vonHoldt et al. 2010, 2011; Schweizer et al. 2016b) and identify the first case of admixture between coastal and NRM wolves in the contiguous US. Wolves from Alaska cluster closely with those from coastal BC (Figs. 2 and 3), which supports previous findings (Weckworth et al. 2005, 2010, 2011; Stronen et al. 2014; Weckworth et al. 2015; Schweizer et al. 2016b; but see Cronin et al. 2014). Our detection of limited differentiation among NRM populations reflects similar findings in vonHoldt et al. (2010). Although to a lesser extent than the coastal/NRM genetic partition, the MT population is distinguishable from the reintroduced populations in ID and YNP and from interior BC and AB. Consequently, the principal genetic partition in the PNW region derives from the coastal and NRM populations.
We assessed the genetic relationships of naturally re-established wolves in WA and OR to potential source populations. Once wolf ancestry was verified using species diagnostic markers, we used evidence from maternal and nuclear markers to identify the source populations’ contributions to the current PNW wolf gene pool. Based on our analyses, the founding WA and OR wolves are migrants from a naturally re-established population in MT, from reintroduced populations in ID and YNP, and for the WA wolves only, from the genetically continuous population in coastal BC and southeast AK (Weckworth et al. 2005; Muñoz Fuentes et al. 2009; Weckworth et al. 2011; Schweizer et al. 2016a, b). Wolves from these source populations may have subsequently admixed within the PNW. An alternative scenario is that founding WA wolves were individuals from previous admixture events of coastal BC and NRM wolves (ID, YNP, MT) that migrated into the state. We find that OR individuals are of NRM ancestry only and find evidence for migrants derived from the YNP/ID cluster in OR. WA individuals have more complex ancestry with some individuals of MT ancestry only and several other individuals with admixed ancestry. These patterns are evident from population assignments within ADMIXTURE and from the presence of several mitochondrial lineages including the lu68 haplotype (Fig. 1, Supplementary Table S1 & S2), which is otherwise known only to exist in coastal wolves. The presence of this haplotype indicates that these individuals are direct migrants either from the coastal population or are offspring of a female wolf with coastal ancestry that dispersed into WA. Migration rates from coastal ecotype into the WA population were estimated to be high as 5% as suggested by results from the BayesAss analysis. However, given that the PCA and ADMIXTURE analyses find mixed nuclear ancestry for these individuals with traces of coastal and NRM wolf ancestry (Figs. 2 and 3, and Supplementary Table S4), it is unlikely they are direct migrants from the coastal population. Limited sampling and high relatedness among some individuals may have reduced our ability to detect migrants and therefore could have led to an underestimate of gene flow occurring between these adjacent populations. Despite these limitations, this study reports the first cases of admixture between coastal and NRM wolves in the contiguous US and illustrates the complex dynamics of admixed populations of conservation concern.
The PNW likely represents an admixture zone between distinct ecotypes for several reasons. First, niche modelling of NRM and coastal wolf distributions indicates that the PNW is an intermediate landscape with environments suitable for both ecotypes in the states of WA and OR (Fig. 4). These results confirm previous findings that the coastal wolf may have extended to southwestern OR or northern California, as supported by the presence of haplotype lu68 as far south as southern OR (Hendricks et al. 2015). Further, as proposed by Young and Goldman (1944), the distribution of C. l. fuscus (the coastal subspecies) extends into these states. Second, wolf packs might create territories in areas that were deemed less suitable environment by the models for both the coastal and NRM populations. Admixed individuals might be well suited to establish in these areas as evident by the Lookout pack in WA. Third, previous research suggests that admixture of wolf subspecies and/or ecotypes can take place over large geographic areas (Schweizer et al. 2016b). Our analyses support this idea, as individuals with coastal ancestry can occupy interior habitat as well as coastal habitat. Fourth, there was a previous absence of wolves in the PNW and there are multiple sources of immigrants in nearby areas. Consequently, admixture between ecotypes in the PNW, as opposed to admixture outside of the PNW with subsequent migration into the PNW, is likely given the diversity of habitats present in the region and the presence of ecotypes in adjacent populations that can provide migrants.
Implications for conservation
The dynamic ancestry of PNW in the future will depend in part on wolf management in western states and the trajectory of population growth in coastal populations. For example, if extreme levels of legalized hunting are practiced in the western US, where the population can be reduced to as few as 150 wolves in each of three western source states (MT, ID, WY; Wayne and Hedrick 2011) and the coastal BC population size remains constant through ongoing protection of the Great Bear Rainforest (BC; Thomson 2016), then the PNW population may continuously receive dispersers with coastal ancestry. On the other hand, if coastal wolves (especially those in the high human impact areas of BC’s south coast and Alaska’s Alexander Archipelago) decline in the future, WA wolves may become a southern refugium that helps safeguard the diversity found in the coastal wolf ecotype.
If genetic influence from the coastal ecotype continues over time, the resulting increase in genetic diversity may allow the population to avoid inbreeding that could lead to the expression of deleterious recessive alleles and cause inbreeding depression as occurred in Scandinavian and Isle Royale wolves (Liberg et al. 2005; Fredrickson et al. 2007; Räikkönen et al. 2009). Although thorough research has yet to be completed, the wolves of the PNW do not show evidence of high levels of inbreeding (here, meaning loss of diversity from a population as measured with the FIS inbreeding coefficient; Supplementary Table S3) or presumed inbreeding depression. Several studies have shown that canids are capable of avoiding mating with close relatives and pack members (Smith et al. 1997a; vonHoldt et al. 2008) through several behavioural mechanisms including absolute avoidance of breeding with related pack members, male-biased dispersal to packs where they breed with nonrelatives and female-biased subordinate breeding. Immigration from other populations will increase the pool of unrelated individuals that can occupy breeding positions or territories. Further, the possible presence of reproductively successful migrants in WA may have influenced genetic diversity. Therefore, the close demographic and genetic monitoring of the population should continue to assess potential inbreeding and inbreeding depression in the PNW populations. Additionally, future projections of the population at carrying capacity should be conducted to determine whether significant inbreeding depression will occur if connectivity and migratory exchange with other populations were to cease (e.g. vonHoldt et al. 2008).
In addition to human-caused mortality, climate change has the potential to negatively affect wolf dynamics and genetic diversity. Theoretical projections suggest that burn areas in WA may increase dramatically (Littell et al. 2010), likely resulting in temporary displacement of prey and, as a result, wolf packs. Further, shifting and reduced habitat of ungulates due to climate change will likely affect the movement of wolves under these scenarios. Although this habitat change may not affect wolf density, it has been shown that disruptions such as human harvest do affect wolf social structure leading to an increase in adoption of unrelated individuals into packs (Rutledge et al. 2010).
Wolf protection and management has led to top–down effects on ecosystem health and function (Berger et al. 2008; Ripple et al. 2015). For example, in YNP, the reintroduction of wolves enhanced restoration of riparian areas, species biodiversity and community complexity (Ripple et al. 2015). Further, wolves often provide other ecosystem and human services such as regulating prey abundance, creating carrion for other species and increasing ecotourism that benefits local economies (Smith et al. 2003; Licht et al. 2010; Ripple et al. 2015; Hendricks et al. 2017).
Complexities of admixture in conservation
Although wolf–coyote hybridization is not common in western North America, introgression of these two species has been found to occur in the American south and Great Lakes area when wolf densities are low and finding a conspecific mate may be difficult (Wayne and Jenks 1991; Lehman et al. 1991; Roy et al. 1994; Koblmüller et al. 2009; vonHoldt et al. 2011, 2016). Given the presence of coyotes in the PNW, individual dispersing wolves or low-density wolf populations, such as those found in western WA, may provide opportunity for coyote–wolf hybridization (see vonHoldt et al. 2011). Even if the coastal ecotype were to become legally protected, wolf–coyote hybrids would not receive protection status due to human influence causing low wolf density resulting in hybridization. Keeping high wolf density and intact pack structure may guard against this possibility and the possibility of wolf–dog hybridization.
While coastal wolf–coyote hybrids would not qualify for protection, coastal wolf–NRM wolf admixed individuals would qualify for protection according to the decision tree criteria presented by Wayne and Shaffer (2016). First, the admixture has resulted between two native populations resulting from natural patterns of wolf dispersal. Second, these admixed individuals are likely ecological surrogates for the coastal wolves and provide similar community interactions and ecosystem functionality. Third, healthy coastal habitats may enhance the proportion of alleles unique to coastal wolves and decrease the fraction of genomic contribution from the NRM (non-endangered) wolf (Wayne and Shaffer 2016). Given their unique evolutionary heritage and adaptations, packs with a dominant coastal ancestry should be considered a priority for conservation.
By providing additional genetic influx to the PNW population, the coastal BC wolf population may enhance adaptation to coastal habitats and enable persistence of wolf populations along the coastal areas. For example, wolves of the coastal ecotype are smaller and focus on salmon and deer as prey rather than larger prey such as elk in NRM populations. They have a unique hunting behaviour for this prey base, including selective eating of salmon parts to avoid parasites and swimming as a means of expanding the deer prey base (Darimont and Paquet 2002; Darimont et al. 2003; Paquet et al. 2006). Currently, there are no established packs within the more coastal areas of the PNW (Fig. 4). Further, allowing for admixture among ecotypes in regions of intermediate habitat may facilitate the process of adaptation and improve the genetic base for selection to act upon (e.g., Hailer and Leonard 2008). As a result, gene flow between coastal BC wolves and NRM populations, such as WA, could potentially help preserve adaptations of the coastal ecotype in an appropriate habitat, enhance the possibility for wolf persistence in coastal habitats of the PNW and enable the evolutionary process of adaptation in intermediate and disturbed habitats. Consequently, we recommend efforts that maintain gene flow and coastal wolf density such as improving and maintaining corridors of immigration and preserving suitable coastal habitat.
Here we provide an example of how managers can use genomic resources to identify ancestry of re-colonized individuals and potential migrants from distinct genetic lineages. Genome-wide analyses are now allowing us to detect signatures of hybridization at a finer scale such as various classes of hybridization such as wolf–dog/wolf–coyote or ecotype–ecotype hybridization, thus advancing our understanding of introgression and divergence. Further, genomic resources (such as the sequence capture methods used here) can be used to inform management decisions as to the most appropriate conservation strategy for a given species (e.g. the distribution of individuals with diagnostic ecotype profiles and their relationship to current and projected habitats). Beyond this study, genomic approaches could be used to identify adaptive potential and further our understanding of preservation of diversity under future climate scenarios (Shafer et al. 2015; Hoffmann et al. 2017).
Policy and management conclusions
Using a multidimensional approach (i.e., combining genomic and ENM analyses to assess admixture during natural re-colonization and the resulting distribution of genetic variation) may offer conservation biologists a methodological approach to discern ecotype admixture zones. These zones, which are often characterized by environmental gradients, provide selective pressure that can contribute to evolutionary change. While in many cases the evolutionary legacy of isolated populations should be preserved, admixture between once-extirpated taxa that has resulted in distinct adaptations should also be considered for protection. Legal protection and conservation guidelines differ depending on the governing body, but many assessments of endangered species policies have recognized the importance of extending some protection to admixed and hybrid populations (Jackiw et al. 2015; vonHoldt et al. 2017). This study, as well as several others (e.g., Weeks et al. 2016; Love Stowell et al. 2017; Frankham et al. 2017), challenges the historical view that admixture and hybridization threaten biodiversity. As advocated by vonHoldt et al. (2017) and Wayne and Shaffer (2016), case-by-case protection should be considered when colonization is a natural process within the integrated WOL framework and when admixed individuals represent effective ecological surrogates that might eventually restore endangered entities to their historical distribution.
Summary
Here we assess admixture during natural re-colonization and the resulting distribution of genetic variation based on mitochondrial haplotypes and 18,508 neutral nuclear SNPs. We utilize niche modelling to define ecotype boundaries and find little correspondence with genetic partitions that may reflect recent colonization from multiple sources. The PNW population is admixed, with coastal influences apparent in WA wolves. This admixture is desirable to enhance adaptation to coastal environments and, in general, enable the evolutionary process for adaptation. Admixed individuals may receive special protection if conditions are such that the historical genetic composition of coastal wolves might be restored and if the hybrids are ecological surrogates providing similar ecosystem functionality and community interactions as the endangered taxon (in this case Alexander Archipelago wolves; see arguments in Wayne and Shaffer (2016)). Determining ecological surrogates may be possible through inferred patterns of selection across the genome, observational studies and/or reciprocal transplant experiments. Further research is needed to establish accurate migration rates and model the potential effects of changing predator/prey dynamics and climate on wolf populations. However, efforts to enhance the density and distribution of coastal wolves in the PNW should be considered as a hedge against population decline in coastal Alaskan or southcoast BC wolves. This effort will aid in the preservation of adaptations for the coastal environment and decrease the likelihood of hybridization with coyotes. To preserve this southern genetic refugium for coastal BC wolves, restore ecological processes and permit contemporary evolution, natural expansion and protection of the coastal wolves in the contiguous US should be an emphasis of wolf management in the PNW.
Data archiving
Sequence reads and mapping files are archived at the NCBI SRA under SRP145376. The filtered variant call file for all individuals as well as a bed file of the neutral regions are available through Dryad Digital Repository under accession number doi:10.5061/dryad.np7t1p2.
References
Abbott RJ, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N et al. (2013) Hybridization and speciation. J Evol Biol 26:229–246
Abbott RJ, Barton NH, Good JM (2016) Genomics of hybridization and its evolutionary consequences. Mol Ecol 25:2325–2332
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Allendorf FW, Leary RF, Spruell P, Wenburg JK (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16:613–622
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arnold ML (2016) Divergence with Genetic Exchange. Oxford University Press. Oxford, UK
Arnold M, Fogarty N (2009) Reticulate evolution and marine organisms: the final frontier? Int J Mol Sci 10:3836–3860
Bailey V (1936) The mammals and life zones of Oregon. North American Fauna No. 55, 272–275.
Berger KM, Gese EM, Berger J (2008) Indirect effects and traditional trophic cascades: a test involving wolves, coyotes, and pronghorn. Ecology 89:818–828
Blair ME, Sterling EJ, Dusch M, Raxworthy CJ, Pearson RG (2013) Ecological divergence and speciation between lemur (Eulemur) sister species in Madagascar. J Evol Biol 26:1790–1801
Boyd DK, Paquet PC, Donelon S, Ream RR, Pletscher DH, White CC (1995) Ecology and conservation of wolves in a changing world. Canadian Circumpolar Institute. Alberta, CA
Callaghan C (2002). The ecology of gray wolf (Canis lupus) habitat use, survival, and persistence in the Central Rocky Mountains, Canada. Carolyn J. Callaghan.
Carmichael LE, Krizan J, Nagy JA, Fuglei E, Dumond M, Johnson D et al. (2007) Historical and ecological determinants of genetic structure in arctic canids. Mol Ecol 16:3466–3483
Chambers SM, Fain SR, Fazio B, Amaral M (2012) An account of the taxonomy of North American wolves from morphological and genetic analyses. North Am Fauna 77:1–67
Chapron G, Kaczensky P, Linnell JDC, Arx von M, Huber D, Andrén H et al. (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519
Cronin MA, Canovas A, Bannasch DL, Oberbauer AM, Medrano JF (2014) Single nucleotide polymorphism (SNP) variation of wolves (Canis lupus) in Southeast Alaska and comparison with wolves, dogs, and coyotes in North America. J Hered 106:26–36
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Darimont CT, Paquet PC (2002) Gray wolves, Canis lupus, of British Columbia's Central and North Coast: distribution and conservation assessment Canadian Field-Naturalist 116:416–422
Darimont CT, Paquet PC, Reimchen TE (2008) Spawning salmon disrupt tight trophic coupling between wolves and ungulate prey in coastal British Columbia. BMC Ecol 8:14
Darimont CT, Reimchen TE, Paquet PC (2003) Foraging behaviour by gray wolves on salmon streams in coastal British Columbia. Can J Zool 81:349–353
DeLong ER, DeLong DM, Clarke-Pearson DL (1988) Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44:837
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498
Diniz-Filho JAF, Bini LM, Rangel TF, Loyola RD, Hof C, Nogués-Bravo D et al. (2009) Partitioning and mapping uncertainties in ensembles of forecasts of species turnover under climate change. Ecography 32:897–906
Dobzhansky T (1935) A critique of the species concept in biology. Philos Sci 2:344–355
Elith J, Graham CH, Anderson RP, Dudík M, Ferrier S, Guisan A et al. (2006) Novel methods improve prediction of species' distributions from occurrence data. Ecography 29:129–151
Elith J, Phillips SJ, Hastie T, Dudík M, Chee YE, Yates CJ (2011) A statistical explanation of MaxEnt for ecologists. Divers Distrib 17:43–57
Faircloth BC, Glenn TC (2012) Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS ONE 7:e42543
Faircloth BC, Sorenson L, Santini F, Alfaro ME (2013) A phylogenomic perspective on the radiation of ray-finned fishes based upon targeted sequencing of ultraconserved elements (UCEs). PLoS ONE 8:e65923
Fourcade Y, Engler JO, Rödder D, Secondi J (2014) Mapping species distributions with MAXENT using a geographically biased sample of presence data: a performance assessment of methods for correcting sampling bias. PLoS ONE 9:e97122
Frankham R, Ballou JD, Ralls K, Dubash MR, Fenster CB, Sunnucks P (2017) Genetic management of fragmented animal and plant populations. Oxford University Press. Oxford, UK
Fredrickson RJ, Siminski P, Woolf M, Hedrick PW (2007) Genetic rescue and inbreeding depression in Mexican wolves. Proc Biol Sci 274:2365–2371
Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM et al. (2014) Genome sequencing highlights the dynamic early history of dogs. PLoS Genet 10:e1004016–12
Fritts SH (1983) Record dispersal by a wolf from Minnesota. J Mammal 64:166–167
Fritts SH, Bangs EE, Fontaine JA, Brewster WG (1995) Restoring wolves to the northern Rocky Mountains of the United States. In: LN Carbyn, SH Fritts, DR Seip (eds) Ecology and conservation of wolves in a changing world. Edmonton, Alberta, p 107–125
Gray MM, Granka JM, Bustamante CD, Sutter NB, Boyko AR, Zhu L et al. (2009) Linkage disequilibrium and demographic history of wild and domestic canids. Genetics 181:1493–1505
Haight RG, Mladenoff DJ, Wydeven AP (1998) Modeling disjunct gray wolf populations in semi‐wild landscapes. Conserv Biol 12:879–888
Hailer F, Leonard JA (2008) Hybridization among three native North American canis species in a region of natural sympatry. PLoS ONE 3:e3333
Harrigan RJ, Thomassen HA, Buermann W, Smith TB (2014) A continental risk assessment of West Nile virus under climate change. Glob Change Biol 20:2417–2425
Harrigan RJ, Thomassen HA, Buermann W, Cummings RF, Kahn ME, Smith TB (2010) Economic conditions predict prevalence of West Nile virus. PLoS ONE 5:e15437
Hedrick PW (2013) Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol 22:4606–4618
Hendricks SA, Charruau PC, Pollinger JP, Callas R, Figura PJ, Wayne RK (2015) Polyphyletic ancestry of historic gray wolves inhabiting U.S. Pacific states. Conserv Genet 16:759–764
Hendricks S, Epstein B, Schonfeld B, Wiench C, Hamede R, Jones ME et al. (2017) Conservation implications of limited genetic diversity and population structure in Tasmanian devils (Sarcophilus harrisii). Conserv Genet 18:977–982
Hernandez PA, Graham CH, Master LL, Albert DL (2006) The effect of sample size and species characteristics on performance of different species distribution modeling methods. Ecography 29:773–785
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Hoffmann AA, Sgrò CM, Kristensen TN (2017) Revisiting adaptive potential, population size, and conservation. Trends Ecol Evol 32:506–517
Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, Luikart G (2011) Next‐generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol Ecol Resour 11:117–122
Jackiw RN, Mandil G, Hager HA (2015) A framework to guide the conservation of species hybrids based on ethical and ecological considerations. Conserv Biol 29:1040–1051
Jimenez MD, Bangs EE, Boyd DK, Smith DW, Becker SA, Ausband DE et al. (2017) Wolf dispersal in the Rocky Mountains, Western United States: 1993–2008. J Wild Mgmt 81:581–592
Jordan MI, Ng AY (2002). On discriminative vs. generative classifiers: a comparison of logistic regression and naive bayes. Adv Neural Information Process Syst. 1-8
Koblmüller S, NORD M, Wayne RK, Leonard JA (2009) Origin and status of the Great Lakes wolf. Mol Ecol 18:2313–2326
Kurtz S, Narechania A, Stein JC, Ware D (2008) A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 9:517
Lehman N, Eisenhawer A, Hansen K, Mech LD, Peterson RO, Gogan PJP et al (1991) Introgression of coyote mitochondrial DNA into sympatric North American gray wolf populations. Evolution 45:104
Leonard JA, Vilà C, Wayne RK (2005) Legacy lost: genetic variability and population size of extirpated US grey wolves (Canis lupus). Mol Ecol 14:9–17
Leonard JA, Wayne RK, Wheeler J, Valadez R, Guillén S, Vilà C (2002) Ancient DNA evidence for Old World origin of New World dogs. Science 298:1613–1616
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Liberg O, Andrén H, Pedersen H-C, Sand H, Sejberg D, Wabakken P et al. (2005) Severe inbreeding depression in a wild wolf (Canis lupus) population. Biol Lett 1:17–20
Licht DS, Millspaugh JJ, Kunkel KE, Kochanny CO, Peterson RO (2010) Using small populations of wolves for ecosystem restoration and stewardship. Bioscience 60:147–153
Littell JS, Oneil EE, McKenzie D, Hicke JA, Lutz JA (2010) Forest ecosystems, disturbance, and climatic change in Washington State, USA. Clim Change 102:129–158
Love Stowell SM, Pinzone CA, Martin AP (2017) Overcoming barriers to active interventions for genetic diversity. Biodivers Conserv 26:1753–1765
Lv W, Li Z, Wu X, Ni W, Qv W (2011) Maximum entropy niche-based modeling (Maxent) of potential geographical distributions of Lobesia botrana (Lepidoptera: Tortricidae) in China. Paper presented at the International Conference on Computer and Computing Technologies in Agriculture V, Beijing, China, 29–31 October, 2011.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M (2010) Robust relationship inference in genome-wide association studies. Bioinformatics 26:2867–2873
Mayr E (1947) Systematics and the origin of species. Columbia University Press. New York, USA
Mech LD (1970) The Wolf. American Museum of Natural History by the Natural History Press. New York, USA
Merrill SB, Mech LD (2000) Details of extensive movements by Minnesota wolves (Canis lupus). USGS Northern Prairie Wildlife Research Center, Paper 76.
Mladenoff DJ, Haight RG, Sickley TA, Wydeven AP (1997) Causes and implications of species restoration in altered ecosystems. Bioscience 47:21–31
Mladenoff DJ, Sickley TA (1998) Assessing potential gray wolf restoration in the Northeastern United States: a spatial prediction of favorable habitat and potential population levels. J Wild Mgmt 62:1
Mladenoff DJ, Sickley TA, Haight RG, Wydeven AP (1995) A regional landscape analysis and prediction of favorable gray wolf habitat in the Northern Great Lakes Region. Conserv Biol 9:279–294
Mladenoff DJ, Sickley TA, Wydeven AP (1999) Predicting gray wolf landscape recolonization: logistic regression models vs. new field data. Ecol Appl 9:37–44
Muhlfeld CC, Kalinowski ST, McMahon TE, Taper ML, Painter S, Leary RF et al (2009) Hybridization rapidly reduces fitness of a native trout in the wild. Biol Lett 5:328–331
Muñoz Fuentes V, Darimont CT, Paquet PC, Leonard JA (2010) The genetic legacy of extirpation and re-colonization in Vancouver Island wolves. Conserv Genet 11:547–556
Muñoz Fuentes V, Darimont CT, Wayne RK, Paquet PC, Leonard JA (2009) Ecological factors drive differentiation in wolves from British Columbia. J Biogeogr 36:1516–1531
Muscarella R, Galante PJ, Soley Guardia M, Boria RA, Kass JM, Uriarte M et al. (2014) ENMeval: an R package for conducting spatially independent evaluations and estimating optimal model complexity for Maxent ecological niche models. Methods Ecol Evol 5:1198–1205
Paquet PC, Alexander SM, Swan PL, Darimont CT (2006) The influence of natural landscape fragmentation and resource availability on connectivity and distribution of marine gray wolf (Canis lupus) populations on Central Coast, BC. Page 726. In: Crooks K, Sanjayan MA (eds) Connectivity Conservation. Cambridge University Press. Cambridge, UK
Paquet PC, Wierczhowski J, Callaghan C (1996) Summary report on the effects of human activity on gray wolves in the Bow River Valley, Banff National Park, Alberta. Pages 74– 120. In: Green J., Pacas, C, Bayley, S, and Cornwell, L (eds) A cumulative effects assessment and futures outlook for the Banff Bow Valley. Department of Canadian Heritage, Ottawa, CA
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Modell 190:231–259
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Räikkönen J, Vucetich JA, Peterson RO, Nelson MP (2009) Congenital bone deformities and the inbred wolves (Canis lupus) of Isle Royale. Biol Conserv 142:1025–1031
Rhymer J, Simberloff D (1996) Extinction by hybridization and introgression. Annu Rev Ecol Syst 27:83–109
Ripple WJ, Beschta RL, Painter LE (2015) Trophic cascades from wolves to alders in Yellowstone. For Ecol Manag 354:254–260
Rodríguez Soto C, Monroy Vilchis O, Maiorano L, Boitani L, Faller JC, Briones MÁ et al. (2011) Predicting potential distribution of the jaguar (Panthera onca) in Mexico: identification of priority areas for conservation. Divers Distrib 17:350–361
Roy MS, Geffen E, Smith D, Ostrander EA, Wayne RK (1994) Patterns of differentiation and hybridization in North American wolflike canids, revealed by analysis of microsatellite loci. Mol Biol Evol 11:553–570
Rödder D, Schmidtlein S, Veith M, Lötters S (2009) Alien invasive slider turtle in unpredicted habitat: a matter of niche shift or of predictors studied? PLoS ONE 4:e7843
Rutledge LY, Patterson BR, Mills KJ, Loveless KM, Murray DL, White BN (2010) Protection from harvesting restores the natural social structure of eastern wolf packs. Biol Conserv 143:332–339
Schweizer RM, Durvasula A, Smith J, Vohr SH, Stahler DR, Galaverni M, Thalmann O, Smith D, Randi E, Ostrander EA, Lohmueller K, Green RE, Novembre J, Wayne RK (2018) Natural selection and origin of a melanistic allele in North American gray wolves. Mol Biol Evol. https://doi.org/10.1093/molbev/msy031
Schweizer RM, Robinson JA, Harrigan RJ, Silva P, Galverni M, Musiani M et al. (2016a) Targeted capture and resequencing of 1040 genes reveal environmentally driven functional variation in grey wolves. Mol Ecol 25:357–379
Schweizer RM, vonHoldt BM, Harrigan RJ, Knowles JC, Musiani M, Coltman D et al. (2016b) Genetic subdivision and candidate genes under selection in North American grey wolves. Mol Ecol 25:380–402
Sesink Clee PR, Abwe EE, Ambahe RD, Anthony NM, Fotso R, Locatelli S et al. (2015) Chimpanzee population structure in Cameroon and Nigeria is associated with habitat variation that may be lost under climate change. BMC Evol Biol 15:2
Shafer ABA, Wolf JBW, Alves PC, Bergström L, Bruford MW, Brännström I et al. (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30(2):78–87. https://doi.org/10.1016/j.tree.2014.11.009
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K et al. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15:1034–1050
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Smith D, Meier T, Geffen E, Mech LD, Burch JW, Adams LG et al. (1997a) Is incest common in gray wolf packs? Behav Ecol 8:384–391
Smith DW, Peterson RO, Houston D (2003) Yellowstone after wolves. Bioscience 53:330–340
Smith TB, Wayne RK, Girman DJ, Bruford MW. (1997b) A role for ecotones in generating rainforest biodiversity. Science 276:1855–1857
Staples J, Nickerson DA, Below JE (2013) Utilizing graph theory to select the largest set of unrelated individuals for genetic analysis. Genet Epidemiol 37:136–141
Stronen AV, Navid EL, Quinn MS, Paquet PC, Bryan HM, Darimont CT (2014) Population genetic structure of gray wolves (Canis lupus) in a marine archipelago suggests island-mainland differentiation consistent with dietary niche. BMC Ecol 14:11
Toppenberg J, Beebe D, Scott, G, Edwards L, Noblin R, Hanson J (2015). Petition to list on an emergency basis the Alexander Archipelago wolf (Canis Lupus Ligoni) as threatened or endangered under the Endangered Species Act. https://www.biologicaldiversity.org/species/mammals/Alexander_Archipelago_wolf/pdfs/Emergency_ESA_petition_for_AA_wolf__14Sep15.pdf
Thomson HS (2016) Bill 2 – 2016- Great Bear Rainforest (Forest Management) Act.
Twyford AD, Ennos RA (2012) Next-generation hybridization and introgression. Heredity 108:179–189
Verts BJ, Carraway LN (1998) Land mammals of Oregon. University of California Press, Berkeley.
Vilà C, Amorim IR, Leonard JA, Posada D, Castroviejo J, Petrucci Fonseca F et al. (1999) Mitochondrial DNA phylogeography and population history of the grey wolf (Canis lupus). Mol Ecol 8:2089–2103
vonHoldt BM, Brzeski KE, Wilcove DS, Rutledge LY (2017) Redefining the role of admixture and genomics in species conservation. Conserv Lett 16:613
vonHoldt BM, Cahill JA, Fan Z, Gronau I, Robinson J, Pollinger JP et al. (2016) Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci Adv 2:e1501714–e1501714
vonHoldt BM, Pollinger JP, Earl DA, Knowles JC, Boyko AR, Parker H et al. (2011) A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res 21:1294–1305
vonHoldt BM, Pollinger JP, Earl DA, Parker HG, Ostrander EA, Wayne RK (2013) Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping. Mamm Genome 24:80–88
vonHoldt BM, Stahler DR, Bangs EE, Smith DW, Jimenez MD, Mack CM et al. (2010) A novel assessment of population structure and gene flow in grey wolf populations of the Northern Rocky Mountains of the UnitedStates. Mol Ecol 19:4412–4427
vonHoldt BM, Stahler DR, Smith DW, Earl DA, Pollinger JP, Wayne RK (2008) The genealogy and genetic viability of reintroduced Yellowstone grey wolves. Mol Ecol 17:252–274
Wall JD, Cox MP, Mendez FL, Woerner A, Severson T, Hammer MF (2008) A novel DNA sequence database for analyzing human demographic history. Genome Research 18:1354–1361
Wayne RK, Hedrick PW (2011) Genetics and wolf conservation in the American West: lessons and challenges. Heredity 107:16–19
Wayne RK, Jenks SM (1991) Mitochondrial DNA analysis implying extensive hybridization of the endangered red wolf (Canis rufus). Nature 351:565–568
Wayne RK, Shaffer HB (2016) Hybridization and endangered species protection in the molecular era. Mol Ecol 25:2680–2689
Weckworth BV, Dawson NG, Talbot SL, Cook JA (2015) Genetic distinctiveness of Alexander Archipelago wolves (Canis lupus ligoni). J Hered 106:412–414
Weckworth BV, Dawson NG, Talbot SL, Flamme MJ, Cook JA (2011) Going coastal: shared evolutionary history between Coastal British Columbia and Southeast Alaska wolves (Canis lupus). PLoS ONE 6:e19582–8
Weckworth BV, Talbot SL, Cook JA (2010) Phylogeography of wolves (Canis lupus) in the Pacific Northwest. J Mammal 91:363–375
Weckworth BV, Talbot S, Sage GK, Person DK, Cook J (2005) A signal for independent coastal and continental histories among North American wolves. Mol Ecol 14:917–931
Weeks AR, Stoklosa J, Hoffmann AA (2016) Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: the case of Australian mammals. Front Zool 13:31
Weir B, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wisz MS, Hijmans RJ, Li J, Peterson AT (2008) Effects of sample size on the performance of species distribution models. Divers Distrib 14:763–773
Wright S (1951) Genetical structure of populations. Annu Eugen 166:247–249
Young S, Goldman EA (1944) The Wolves of North America, vols 1 and 2. American Wildlife Institute, Washington.
Zhang W, Fan Z, Han E, Hou R, Zhang L, Galaverni M et al (2014) Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai-Tibet Plateau. PLoS Genet 10:e1004466–13
Acknowledgements
We are grateful to Kristin Mansfield and Roblyn Brown for samples and insight into Washington and Oregon wolves, respectively. Laboratory and data processing assistance was completed by Sarah Wenner, Daniel Greenfield and Jacqueline Robinson. We thank Brendan Epstein for helpful discussions concerning methods' implementation and Kim Andrews for discussions regarding data interpretation within this study. This work was supported by the Wilburforce Foundation and the National Science Foundation (NSF; DEB-1021397, OPP-0733033). Additional support was provided to SAH by the National Institute of Health (P30GM103324), NSF (DEB-1316549) and the Bioinformatics and Computational Biology program at the University of Idaho; to RMS by the NSF (DGE-1144087, DGE-0707424, 1612859); and to RJH by the NSF (PD-08-1269). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH (S10RR029668, S10RR027303).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hendricks, S.A., Schweizer, R.M., Harrigan, R.J. et al. Natural re-colonization and admixture of wolves (Canis lupus) in the US Pacific Northwest: challenges for the protection and management of rare and endangered taxa. Heredity 122, 133–149 (2019). https://doi.org/10.1038/s41437-018-0094-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-018-0094-x
This article is cited by
-
Development of a mitochondrial DNA marker that distinguishes domestic dogs from Washington state gray wolves
Conservation Genetics Resources (2020)
-
Conservation genomics illuminates the adaptive uniqueness of North American gray wolves
Conservation Genetics (2019)
