
Abstract
Purpose
Making a diagnosis from clinical genomic sequencing requires well-structured phenotypic data to guide genotype interpretation. A patient’s phenotypic features can be documented using the Human Phenotype Ontology (HPO), generating terms used to prioritize genes potentially causing the patient’s disease. We have developed GenomeDiver to provide a user interface for clinicians that allows more effective collaboration with the clinical diagnostic laboratory, with the goal of improving the success of the diagnostic process.
Methods
GenomeDiver uses genomic data to prompt reverse phenotyping of patients undergoing genetic testing, enriching the amount and quality of structured phenotype data for the diagnostic laboratory, and helping clinicians to explore and flag diseases potentially causing their patient’s presentation.
Results
We show how GenomeDiver communicates the clinician’s informed insights to the diagnostic lab in the form of HPO terms for interpretation of genomic sequencing data. We describe our user-driven design process, the engineering of the software for efficiency, security and portability, and examples of the performance of GenomeDiver using genomic testing data.
Conclusion
GenomeDiver is a first step in a new approach to genomic diagnostics that enhances laboratory–clinician interactions, with the goal of directly engaging clinicians to improve the outcome of genomic diagnostic testing.
Graphical Abstract

Similar content being viewed by others

INTRODUCTION
Compelling economic evidence now supports how genomic sequencing saves both money and time1,2 and improves quality-adjusted life years3 in the diagnosis of the 3.5–5.9% of the population with rare diseases.4 To make this genomic diagnostic process more efficient, substantial attention has been appropriately paid to detecting and understanding the effects of sequence variants in the human genome.5 Linking genetic variants to diseases is challenging and is most successful in case–control studies or when comparing multiple affected and unaffected family members, for which excellent analytical approaches have been developed.6,7 In a clinical context, however, testing can only be ordered on the proband and one or both parents (if available), with results needed sufficiently rapidly to influence care. The genomic diagnostic laboratory relies on linking the variants present in the patient’s genome with the patient’s phenotypic features. When a pathogenic genetic variant has previously been found to be the cause of a disease, that associated disease is characterized by a set of Human Phenotype Ontology (HPO) terms.8 If the patient has a variant that appears potentially pathogenic, causality is supported if the list of HPO terms in the disease associated with a pathogenic variant of that gene significantly overlaps the HPO terms used to describe the patient. A number of phenotype-based variant prediction tools exist to perform this kind of variant prioritization.9
If the diagnostic laboratory is provided with a greater number of HPO terms describing the patient on whom sequencing was performed, the diagnostic yield is increased.10,11 The comprehensive collection of these HPO terms is very difficult to scale effectively. Ordering genomic tests for rare diseases involves providing the lab with a primary indication for the testing (e.g., hearing loss, seizures), but also requires communicating additional findings in the patient, sometimes through checklists completed as part of the test order, also potentially involving the diagnostic laboratory reviewing notes from the patient’s health record. The diagnostic laboratory staff then extract information from these sources and make decisions about how the phenotype can be represented as HPO terms. In critically ill children, an approach has been described that involves automated extraction of HPO terms from the electronic health record (EHR) using natural language processing,12 helping with the speed of generation of HPO terms.
We have developed GenomeDiver as part of the NYCKidSeq project13 to help clinicians contribute more effectively to the diagnostic process. The NYCKidSeq project is jointly funded by the National Human Genome Research Institute and the National Institute on Minority Health and Health Disparities, and is one of seven national clinical projects that are part of the Clinical Sequencing and Evidence-generating Research Consortium (CSER).14 GenomeDiver facilitates the curation of HPO terms by the clinician quickly, in greater numbers and more accurately than is currently typical, with the goal of improving our ability to make diagnoses using genomic information.
MATERIALS AND METHODS
The user-centered design process for GenomeDiver
We developed GenomeDiver using evidence-based participatory methods from design thinking.15 User research began with initial interviews with physicians and genetic testing laboratory staff, followed by a one-day stakeholder workshop and a design sprint to understand user needs, to define requirements, to sketch candidate workflows, to choose among them and to prototype. We included physicians (with and without specialist genetics training), genetic counselors, genetic testing laboratory personnel, research scientists, software tool builders, bioinformaticians and biologists, a designer, and a software engineer. The resulting concepts, sketches, and paper prototypes informed the designs for user experience, workflow, and user interfaces.
The GenomeDiver workflow: overview of a “dive”
The workflow for a case (Figure S1) begins with the upload by the genetic testing lab of the patient’s genomic information as a variant call format (VCF) file, along with the HPO terms that they derived from the test requisition documents. The clinician then starts a “dive” by selecting the patient from the GenomeDiver interface. The next step, refining the patient’s phenotype, requires the clinician categorize HPO terms generated by GenomeDiver by dragging each into one of three screen areas: “Present” in the patient, “Absent,” or “Unknown.” Using this updated and enriched phenotypic information, GenomeDiver reanalyzes the genomic data and presents candidate genes and associated diseases for the clinician to review and flag, adding any comments that they would like to communicate to the diagnostic laboratory. To conclude the workflow, the updated HPO term categorizations, flagged diseases, clinician comments, and the reranked shortlist of variants are returned to the diagnostic laboratory for interpretation.
Variant prioritization and reverse phenotyping to select HPO terms
We illustrate the variant prioritization steps in Figure S2. The VCF and starting HPO information are used for an Exomiser16 analysis. Exomiser is a gene prioritization tool that combines a score quantifying the likely pathogenicity of a variant associated with a gene (variant score) with a second score that measures the similarity of the phenotypic features of the patient with those associated with a pathogenic variant of the gene (phenotype score). Both values are used to generate the combined score for the variant. GenomeDiver interacts with Exomiser by using Exomiser as a source of prioritized genes, and by feeding back enhanced HPO information to help Exomiser refine its predictions.
We describe the prioritization and filtering steps in detail in the Supplementary Information. An Exomiser run is initiated based on the patient’s VCF and the HPO terms from the test requisition. This generates a list of variants, each of which is associated with a gene. Each gene in turn is associated with one or more diseases, and these individual diseases are described by sets of HPO terms. We collect the HPO terms for the genes that Exomiser has ranked highest for causing the patient’s phenotype. These are filtered for redundancy, focusing on those that are descendants of “Phenotypic abnormality” (HP: 0000118), and those associated with the specific disease prioritized by Exomiser. This leaves a subset of HPO terms that go through multiple rounds of selection, with the goal to present to the clinician ≤5 nonredundant HPO terms associated with each candidate gene, generating a maximum of 25 new HPO terms for categorization.
Reanalysis based on enriched HPO information and disease exploration
A further potential input by the clinician is made possible by rerunning Exomiser, now updated with the newly categorized HPO terms that are used by Exomiser to generate revised combined scores. GenomeDiver then presents the clinician with a list of genes ranked by the absolute combined score, displaying the magnitude and direction of change of the scores, and linking to associated candidate diseases. The hyperlink embedded with the names of each candidate disease brings the clinician to a description of the disease, letting them judge whether the syndrome of features plausibly fits their patient. Any disease of interest can be flagged, with the final step of the dive involving the clinician returning the list of updated HPO terms, any flagged diseases and free text comments as a file for the diagnostic laboratory.
User experience trial
We performed a user experience trial of the software with four of the NYCKidSeq clinicians, none of whom had used the software previously. Six NYCKidSeq patients lacking a final genomic diagnosis were selected, and the clinical synopsis generated on each from medical records was provided to each clinician. An initial user survey was performed to learn about prior GenomeDiver experience and current practice when communicating with genomic diagnostic laboratories. The time spent at each stage of GenomeDiver interactions was recorded, followed by an exit survey, in which users were asked to evaluate their experience.
Software system architecture
We provide a detailed description of the GenomeDiver software in the Supplementary Information.
RESULTS
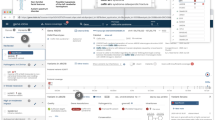
GenomeDiver performance with simulated data
A video of the diagnostic laboratory interaction with the interface to set up a patient in the GenomeDiver system appears as Supplementary Video 1. Once set up, this allows a clinician to access the separate interface shown in Fig. 1 for categorization of HPO terms. The upper section “Add Phenotype Feature” allows text to be entered, prompting HPO terms as a dropdown list for selection by clicking. The design is intended to focus the clinician on the middle section to “Classify All Phenotype Features,” dragging and dropping individual HPO terms into categories of “Present,” “Absent,” or “Unknown.” After all terms have been categorized, the “Submit” button on the bottom right activates and the updated information is sent back to GenomeDiver. This interaction appears in Supplementary Video 2.

Human Phenotype Ontology (HPO) terms can be added in the “Add Phenotype Feature” field by entering text that will bring up a list of HPO terms from which the appropriate term can be chosen. The HPO terms generated by GenomeDiver are shown in the “Classify all phenotype features” section. These can be dragged into each of the categories underneath, “Present,” “Absent,” or “Unknown.” Three terms have been moved into the “Present” category as an example.
We used the VCF for the publicly available NA12878 genome,17 adding a variant of uncertain significance in the FBN1 gene (ClinVar accession VCV000200085.4, NM_000138.4[FBN1]: c.6449G>T[p.Arg2150Leu]). To simulate a request for analysis of a patient presenting with a clinical suspicion of Marfan syndrome (OMIM 154700), we used HPO terms for ascending tubular aorta aneurysm (HP: 0004970), scoliosis (HP: 0002650), and arachnodactyly (HP: 0001166) as those potentially used in a test requisition in a clinical scenario.
We detail each step of the process of variant and gene filtering and prioritization in the Supplementary Information section (Figure S2), as well as the selection of HPO terms for presentation to the clinician (Figures S3–S4). For this particular combination of genomic and phenotypic information, two genes were selected, FBN1 and SKI, the latter implicated in Shprintzen–Goldberg syndrome (OMIM 182212), whose phenotypic features likewise include aortic aneurysm, scoliosis, and arachnodactyly.18 The HPO terms presented to the clinician are listed in Supplementary Table 1. We show how each term in our sample case was categorized in Supplementary Table 1 and illustrate how a second Exomiser run presents candidate diseases in Fig. 2 (using steps illustrated in Figure S5). Of note, GenomeDiver never exposes information about variants to the clinician, preventing the clinician from bypassing the formal laboratory diagnostic reporting process.

The clinician is presented with a shortlist of genes, ranked by the Exomiser combined_score, which is shown, as well as the change in score (green positive, red negative) resulting from the HPO term categorization. We list the disease and a link that allows the clinician to explore whether a disease description resembles their patient, allowing them to flag one or more as being candidates for causing the patient’s presentation. Free text can be used in the “Add comment” box, finalizing the process by clicking “Confirm analysis” to return the information to the diagnostic laboratory.
User experience trial results
The time spent categorizing HPO terms is shown in Figure S6. The median time spent in categorizing HPO terms across the six patients was 203 seconds (3 minutes and 23 seconds). Categorization tended to be concordant between users (Figure S7). The subsequent interaction evaluating candidate genes and diseases took 134.5 seconds (2 minutes and 14.5 seconds) per case (Figure S8). Exit survey data revealed overall positive responses to the use of GenomeDiver, particularly for interface intuitiveness, speed, and satisfaction in contributing to the diagnostic process (Figure S9), with all users reporting that they could foresee using GenomeDiver in their clinical practice.
DISCUSSION
In this initial deployment of GenomeDiver, we have focused on improving the diagnostic process when performing genome-wide (exome, genome) sequencing in the diagnosis of rare diseases. We recognize that we can potentially improve both the rate of successful diagnosis and decrease the time spent by diagnostic laboratory personnel if we can facilitate the provision of the HPO terms that are most likely to discriminate the highest ranked gene candidates for causing the patient’s disease. With the appreciation that clinician time is limited, the interface is designed to be simple and intuitive, limiting the number of candidate HPO terms so that the entire categorization session lasts no more than a few minutes per patient, validated by our user experience pilot testing. The second stage of input, facilitating the exploration of diseases that could be affecting the patient, permits further clinician insights to be provided, this time studying the syndrome of phenotypic features in candidate diseases. These steps of identifying individual phenotypic features and then considering how they might aggregate in diseases and syndromes reflect reasonably accurately the typical clinical genetics evaluation, but with the added intent of contributing to the laboratory diagnostic process. While we present our choices for variant and HPO prioritization in the current version, these are intended to serve as the basis for the initial, functional version of GenomeDiver, but with the goal of incorporating community input for algorithmic improvement over time.
It is likely that HPO term harvesting from sources like EHRs19 and image analysis20 represents an area of innovation that will expand over time. A value of the GenomeDiver interface is that it allows a clinician to assess whether candidate terms gleaned from diverse, automated sources are actually present in a patient, creating the potential that GenomeDiver can act as a clearinghouse for downstream curation of candidate phenotypic features. The categorization of HPO terms as “absent” is not yet used by Exomiser, but the updated LIRICAL approach, based on a likelihood ratio framework that includes information about HPO prevalence data,21 can exploit such information and will be served by this functionality within GenomeDiver.
Several lessons from the user experience trial will be valuable for guiding ongoing development of GenomeDiver. Manual addition of HPO terms was an option chosen by one of the users, associated with significantly longer interaction with the software. As this is an option, not a requirement for progressing with a dive, this decision is at the discretion of the user, but it would be beneficial to make it more time-efficient. The HPO term harvesting described above is likely to overcome some of the challenges of manual curation, but will increase the number of HPO terms for categorization, which was correlated with a prolonged duration of time interacting with the software (Figure S6). To address this, we will need to provide a way of presenting or prioritizing terms with the highest information content, and consider interface improvements to allow even more rapid categorization of terms. We also need to understand nonconcordance between users of HPO term categorization. On a positive note, we find it reassuring that the software does not constrain users into uniform decisions. With more use in a production setting, we will be able to study how interuser variation is associated with success in diagnostic outcomes, allowing us to identify less productive interactions with the software, which can in turn prompt redesign to promote its more effective use.
GenomeDiver may find uses beyond initial diagnoses of patients with a rare disease. GenomeDiver can facilitate reanalysis of patients with initially uninformative results whose phenotypes may have changed over time, and whose genomic variants may have undergone reclassification. GenomeDiver is fundamentally a tool to prompt “reverse phenotyping”—checking whether specific phenotypic features are present in a patient based on their genotype. As such, GenomeDiver can serve clinicians not trained in clinical genetics or dysmorphology. With the development of HPO terms that now also encompass common diseases,22 GenomeDiver’s potential value as a reverse phenotyping tool that generates a genotype-based differential diagnosis could accordingly extend to much of medical genomics.
Data availability
The GenomeDiver software is available at https://github.com/GenomeDiver/. The patient genomes and HPO terms used in the user experience trial will be available as part of the Clinical Sequencing Evidence-Generating Research (CSER) data deposition on the Analysis, Visualization, and Informatics Lab-space (ANViL, anvilproject.org).
References
Splinter, K. et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379, 2131–2139 (2018).
Chung, C. C. Y. et al. Rapid whole-exome sequencing facilitates precision medicine in paediatric rare disease patients and reduces healthcare costs. Lancet Regional Health Western Pacific. 1, 100001 (2020).
Schofield, D., Rynehart, L., Shresthra, R., White, S. M. & Stark, Z. Long-term economic impacts of exome sequencing for suspected monogenic disorders: diagnosis, management, and reproductive outcomes. Genet. Med. 21, 2586–2593 (2019).
Nguengang Wakap, S. et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173 (2020).
Rehm, H. L. & Fowler, D. M. Keeping up with the genomes: scaling genomic variant interpretation. Genome Med 12, 5 (2019).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Hu, H. et al. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet. Epidemiol. 37, 622–634 (2013).
Köhler, S. et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res 47, D1018–D1027 (2019).
Pengelly, R. J. et al. Evaluating phenotype-driven approaches for genetic diagnoses from exomes in a clinical setting. Sci. Rep. 7, 13509 (2017).
Trujillano, D. et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182 (2017).
Thompson, R. et al. Increasing phenotypic annotation improves the diagnostic rate of exome sequencing in a rare neuromuscular disorder. Hum. Mutat. 40, 1797–1812 (2019).
Clark, M. M. et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 11, eaat6177 (2019).
Odgis, J. A. et al. The NYCKidSeq project: study protocol for a randomized controlled trial incorporating genomics into the clinical care of diverse New York City children. Trials 22, 56 (2021).
Amendola, L. M. et al. The Clinical Sequencing Evidence-Generating Research Consortium: integrating genomic sequencing in diverse and medically underserved populations. Am. J. Hum. Genet. 103, 319–327 (2018).
Brown, T. Design thinking. Harvard Bus. Rev. 86, 84–92 (2008).
Smedley, D. et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 10, 2004–2015 (2015).
Zook, J. M. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data. 3, 160025 (2016).
Greally, M. T. Shprintzen-Goldberg Syndrome. in GeneReviews® (eds Adam, M. P. et al.) (University of Washington, Seattle, 1993).
Deisseroth, C. A. et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet. Med. 21, 1585–1593 (2019).
Hsieh, T.-C. et al. PEDIA: prioritization of exome data by image analysis. Genet. Med. 21, 2807–2814 (2019).
Robinson, P. N. et al. Interpretable clinical genomics with a likelihood ratio paradigm. Am. J. Hum. Genet. 107, 403–417 (2020).
Groza, T. et al. The Human Phenotype Ontology: semantic unification of common and rare disease. Am. J. Hum. Genet. 97, 111–124 (2015).
Acknowledgements
Research reported in this publication was part of the NYCKidSeq project, supported by the National Human Genome Research Institute and National Institute for Minority Heath and Health Disparities of the National Institutes of Health under award number 1U01HG0096108.
Author information
Authors and Affiliations
Contributions
Conceptualization: N.M.P., C.S., K.S., F.B., N.S.A-H., K.B., J.A.O., S.A.S., B.D.G., E.E.K., V.J., T.B., J.M.G. Data curation: V.J. Formal analysis: N.M.P., F.B., C.G., T.B., J.M.G. Funding acquisition: C.R.H., M.W., B.D.G., E.E.K. Investigation: N.M.P., C.S., K.S., F.B., T.B., J.M.G. Methodology: N.M.P., C.S., K.S., F.B., C.G., V.J., T.B., J.M.G. Project administration: N.M.P., F.B., G.B., E.E.K., C.G., V.J., T.B., J.M.G. Resources: C.R.H., M.W., B.D.G., E.E.K., C.G., V.J., T.B., J.M.G. Software: N.M.P., C.S., K.S., F.B., J.M.G. Supervision: G.B., E.E.K., C.G., J.M.G. Validation: N.M.P., C.S., K.S., F.B., N.S.A-H., G.A.D., M.W., B.D.G. Visualization: N.P., C.S., K.S., F.B., J.M.G. Writing—original draft: N.M.P., CS KS C.G., J.M.G. Writing—review & editing: N.M.P., C.S., K.S., F.B., N.S.A-H., GB K.B., G.A.D., J.A.O., S.A.S., C.R.H., M.W., B.D.G., E.E.K., C.G., T.B., J.M.G.
Corresponding author
Ethics declarations
Ethics declaration
The NYCKidSeq project (ClinicalTrials.gov Identifier: NCT03738098) was approved by the Institutional Review Boards (IRBs) of Icahn School of Medicine at Mount Sinai and Albert Einstein College of Medicine. Informed consent was obtained from all study participants. All data were de-identified for downstream research, including the GenomeDiver user experience trial.
Competing interests
N.S.A.-H. was previously employed at Regeneron Pharmaceuticals and has received an honorarium from Genentech. E.E.K. has received speaker honoraria from Regeneron Pharmaceuticals and Illumina, Inc. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pearson, N.M., Stolte, C., Shi, K. et al. GenomeDiver: a platform for phenotype-guided medical genomic diagnosis. Genet Med 23, 1998–2002 (2021). https://doi.org/10.1038/s41436-021-01219-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01219-5
This article is cited by
-
Predicting genes from phenotypes using human phenotype ontology (HPO) terms
Human Genetics (2022)

{kind=link}