
Abstract
Purpose
Genomic medicine holds great promise for improving health care, but integrating searchable and actionable genetic data into electronic health records (EHRs) remains a challenge. Here we describe Neptune, a system for managing the interaction between a clinical laboratory and an EHR system during the clinical reporting process.
Methods
We developed Neptune and applied it to two clinical sequencing projects that required report customization, variant reanalysis, and EHR integration.
Results
Neptune has been applied for the generation and delivery of over 15,000 clinical genomic reports. This work spans two clinical tests based on targeted gene panels that contain 68 and 153 genes respectively. These projects demanded customizable clinical reports that contained a variety of genetic data types including single-nucleotide variants (SNVs), copy-number variants (CNVs), pharmacogenomics, and polygenic risk scores. Two variant reanalysis activities were also supported, highlighting this important workflow.
Conclusion
Methods are needed for delivering structured genetic data to EHRs. This need extends beyond developing data formats to providing infrastructure that manages the reporting process itself. Neptune was successfully applied on two high-throughput clinical sequencing projects to build and deliver clinical reports to EHR systems. The software is open source and available at https://gitlab.com/bcm-hgsc/neptune.
Similar content being viewed by others
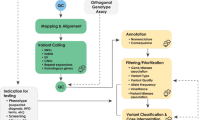

INTRODUCTION
Genomic medicine seeks to improve clinical outcomes by identifying risk for adverse drug events, providing molecular diagnoses, and identifying patients with increased lifetime risk of genetic disease, but implementation is limited by many factors. These include (1) insufficient infrastructure for high-throughput clinical reporting [1,2,3], (2) challenges handling protected health information (PHI) [4, 5], (3) labor-intensive genomic variant interpretation [6], (4) clinical site specific data integration requirements [7, 8], (5) few actionable findings in some disease areas [9], (6) additional burden on providers to integrate genetic data [10], and (7) a reluctance from insurance providers to pay for precision medicine testing [11]. Addressing these challenges demands research that pairs large genomic data sets with clinical outcomes. Many national and international clinical sequencing projects have been established to fill this need, including the eMERGE Network [12], All of Us [13], the IGNITE network [14], and the Clinical Sequencing Evidence-Generating Research [15] (CSER) consortium as well as a large number of private and regional initiatives [16, 17].
Integrating genomic data in electronic health records (EHRs) will allow researchers to improve the clinical impact of genomic data, demonstrate its utility, and make it accessible to clinical decision support tools. Unfortunately, genomic data are often heterogeneous, mix or lack standards, are updated regularly, and require domain expertise to handle correctly. Data standards are in development [18] but there is a lack of flexible, comprehensive, and open source solutions for structuring genomic data and cleanly bridging the gap to EHR systems. There are commercial clinical reporting offerings in this space, but they are closed source [19,20,21]. PharmCat combines a similar set of features by capturing domain knowledge, providing sample analysis, and generating clinical reports, but it focuses on pharmacogenomic reporting [22]. Genomics informatics resources like dbGaP [23] offer longer-term data storage solutions or focus on reanalysis [24]. Finally, some tools provide general support for building HL7 or FHIR messages, but do not provide domain-specific support for clinical genetics reporting [25]. In summary, Neptune offers a robust, open source package of tools for integrating genomics data into the EHR (Supplementary Table 1).
While many laboratories have solutions to aid variant interpretation, incorporating structured genetic testing results into the EHR is widely considered so difficult that few laboratories attempt it, instead preferring to load PDFs as media files [26]. To support delivering genomic data to the EHR, we have developed Neptune, an environment that manages the clinical reporting process. The key features of Neptune are (1) to take as input genomic data (genotypes and coverage information) and compare against a VIP database of known genetic variation, marking known variants with previously curated data, selecting novel genomic variants for review, and identifying samples where all variants have been curated, which is essential for automated reporting; (2) to combine data from diverse sources including sample metadata from a Laboratory Information Management System (LIMS) and variant information from the VIP database and output data in a structured report file ready to be accepted by EHR systems; (3) to convert that structured data into a customizable human-readable report; (4) to enable corrected and updated reports; and (5) to enable the reanalysis and reinterpretation of data over time. In this report we describe Neptune’s workflow and its application to two gene panel based clinical tests that required data integration into EHRs: eMERGE III and HeartCare.
MATERIALS AND METHODS
Following the detection of genomic variants using standard bioinformatics pipelines [27], Neptune communicates via API with an external variant interpretation interface to obtain the most up-to-date valiant interpretation data. Annotated variants and associated metadata are used to populate a structured JSON format that represents the clinical report for that sample. This functionality is encapsulated in an API (Table 1). Automated reporting is possible when all variants in a sample have been previously curated.
VIP database
The VIP database of genomic variation is maintained externally from Neptune. This database contains variant information (position, allele), frequency, transcript data, gene annotations (disease association, inheritance), and internal curation data (PubMed IDs of related publications; comments and categories from clinical sites). It currently contains data from 381,564 variants (Fig. 1b). This database was initially seeded by the two clinical reporting laboratories for the eMERGE III network [28], and has been subsequently updated to incorporate novel variants that are detected in samples in the Baylor College of Medicine Human Genome Sequencing Center (HGSC) Clinical Lab and other public variant resources. This resource draws on both public resources (ClinVar, OMIM, literature review) and internal data sets. The VIP database is available for download at https://gitlab.com/bcm-hgsc/neptune. Neptune interacts with a snapshot of the VIP database in vcf format. If a clinical laboratory maintains its own variant database, Neptune can be modified to retrieve it instead using Neptune’s module system, or the ClinVar data format could be used directly.

(a) Neptune manages the variant review process and brings together disparate data from multiple external systems to create a final report file, in either JSON, HTML, or FHIR format. Central to this process is the VIP database of genetic variation. For each sample, novel genomic variants are added to this database and curated as needed according to project-specific rules. (b) The contents of the VIP database include curated variants. VIP database variants are predominantly variants of uncertain significance (VUS) or likely benign. CNV copy-number variant, EMR electronic medical record.
Variant filtering and interpretation in eMERGE and HeartCare
Clinical genomic variant filtering and interpretation were implemented separately from Neptune in an annotation pipeline and external curation interface, following American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines. As ClinGen recommendations become available (e.g., MYH7 [29] or copy-number variant (CNV) guidelines [30]) we have adopted them. eMERGE and HeartCare used a similar set of project-specific filters to reduce the review burden of benign variation. These filters were implemented separately and are not part of Neptune.
To calculate precision, recall, f measure, and specificity, we define a positive as a reportable, pathogenic variant and not reportable variants as negative. A true positive then would be a reportable variant was either in the VIP or novel (i.e., was selected for review), a false positive would be a variant selected for review that was not reportable, and a false negative would be a variant that was reportable that was not selected for review. Metrics were evaluated for a recent batch (IR277) containing 138 samples.
Variant annotation with locally curated variant data
Novel variants are detected by comparing their genomic coordinates and alternate allele. Variants that are not present in the VIP database can be forwarded to a variant review system for manual curation. Following manual curation, novel variants are added to the VIP database by an external tool. Once all variants in a sample have been categorized, Neptune extracts reportable, pathogenic variants using curations stored in the VIP database, and outputs an automated clinical report populated with prioritized variants (or a negative report if no relevant variants are found).
The assessment of variants reviewed per sample in this study (Fig. 2) was done by “replaying” our review process, starting from an empty VIP database. Variants were limited to the 68 eMERGE consensus reportable genes (Supplementary Table 1). Each sample was analyzed in the order in which it was received. For each variant selected for review during our initial review process, we checked for it in the database. The database was empty or nearly empty early in this process, so many variants were assessed. We then added all reviewed variants to the database. As we progressed through the 7,258 data freeze samples we recorded how many reviewable variants were not present in the database for each new sample.

The plot shows the number of variants per sample requiring review in 68 eMERGE III consensus reportable genes, starting with an empty database. As additional samples are reviewed from a data freeze of 7,258, the number of variants per sample that are selected quickly decreases. In eMERGE III, the number of variants that require review plateaus at around one variant per sample.
Copy-number variation
Neptune can integrate CNVs by incorporating AtlasCNV [31] output into the report. If activated, reports contain a CNV section. CNVs and single-nucleotide variants (SNVs) are reported alongside one another to highlight cases of compound heterozygosity, in which one gene contains both a CNV and another deleterious variant. Many of the CNVs reported in these studies were reviewed prior to the release of guidelines by ClinGen [30, 32], though reviews conducted after their release followed them. Prior to their release we applied ClinGen haploinsufficiency/triplosensitivity data, assessed whether the CNV was in or out of frame if possible, and considered known pathogenic CNVs or indels that overlapped the CNV in question. In eMERGE we initially required the CNV to span three exons until the release of our updated CNV caller, atlas-CNV [31] which allowed us to begin reporting single-exon CNVs. In HeartCare, we reported single-exon events throughout the duration of the project.
Pharmacogenomics
Pharmacogenomic analysis is available for a subset of commonly reported genotypes and star alleles [33]. The module is configurable and the set of pharmacogenomic findings that are reported are defined using a mapping file that links reportable genotypes to their associated star alleles, phenotypes, and interpretation notes. Pharmacogenomic analysis requires either a gvcf input or external QC file with coverage values for all pharmacogenomic variant sites. Variants are assumed to be unphased, leading to ambiguous star allele assignments in some cases (e.g., TPMT *1/*3A vs. *3b/*3c). If the pharmacogenomic analysis is active, an additional table will be added to the report that describes the pharmacogenomic variants in the patient, as well as adding the corresponding data to the structured JSON file.
Polygenic risk scores
Neptune includes a module that enables the clinical reporting of polygenic risk score (PRS). This module reads a file in variant call format (vcf), restricted to sites of interest for a given PRS. It then calculates the risk score, using weights provided in a configuration file and the zygosity of each allele. Finally, the score for each sample is then compared against a reference distribution (also provided in the configuration) to determine the risk category for that sample. The PRS, risk category, and weighted genotypes can be added to structured outputs. Although the clinical utility of PRS is currently not settled [32], gathering additional clinical data sets will facilitate the assessment of their utility.
Report templates
Reports are designed to meet all CAP/CLIA requirements and are highly customizable using an HTML-based templating system. Sections of the report can be activated or deactivated based on sample metadata such as project or sequencing methodology. Neptune supports both corrections and amendments to existing reports, with changes tracked and timestamped. By integrating with our variant review system, our internal deployment of Neptune streamlines the generation of batches of negative reports, which is critical in projects with a large number of negative reports.
Conversion to structured data formats
Neptune allows structured outputs to be in one of a variety of formats, including FHIR, HTML, and JSON. Regardless of the format, the output captures all elements of the report including variant information, descriptive text, and coverage statistics produced by the ExCiD software. In the next step, this prereport is merged with PHI within a fully HIPAA-compliant environment and the final report is made available to a laboratory director for approval. For ease of viewing, an HTML version of the report is also made available.
For the eMERGE III project, the JSON file was converted into a proprietary XML format selected for use by the eMERGE network. This format was standardized across the two clinical reporting laboratories which allowed clinical sites to accept reports in a unified format [34]. In our HeartCare project, work is ongoing to develop a FHIR-compatible data specification and a conversion tool that can take this specification and JSON data to produce FHIR-compatible outputs (https://emerge-fhir-spec.readthedocs.io/en/latest/).
The BCM HeartCare study
In the Baylor College of Medicine (BCM) HeartCare study, patients who presented at BCM clinical sites were invited to participate in a clinical genomics study that included return of genomic results and integration into the EHR. This project increased the complexity of the clinical report by adding a section for reporting a PRS alongside integrated small variant and CNV genomic findings from 168 genes related to cardiac disease, pharmacogenomic findings for a set of drugs related to cardiovascular disease, and the reporting of two risk alleles [35] for lipoprotein(a) (Lp(a)) [36].
RESULTS
We developed Neptune to facilitate delivering genetic test data to EHRs. Neptune follows object oriented design principles, with separate classes used to contain logic for samples, metadata, variants, VIP snapshots, report builders, and database connections among others (Supplementary figure 1). A key challenge with developing a system like Neptune is separating logic that is specific to the clinical laboratory in which it was developed from generalizable logic. To address this, we created a module system that allows development of separable components. These modules are loaded dynamically, based on a configuration file. For example, the report for a particular project may include CNVs, so the CNV “report_feature” can be activated in that project’s configuration file, which will instruct Neptune on the module to use for loading and displaying CNVs on the report. Neptune depends on the pyyaml, qrcode, and sqlite3 python packages. The FHIR client is also developed at the HGSC and available at https://gitlab.com/HGSC-NGSI/heartcare/heartcare-hl7.
Case study: Electronic Medical Records and Genomics Network
The eMERGE Network brings together researchers and clinical laboratories to study the implementation of genomic medicine [28]. Previously, as part of the eMERGE III Network, we performed clinical interpretation and issued over 14,500 clinical reports to 7 clinical sites for a targeted gene panel of 68 consensus genes with additional clinical site specific genes. Clinical reports needed to be customized to each clinical site, which presented a challenge. Customizations included modifying the gene list depending on the clinical site, allowing specific single-nucleotide polymorphisms (SNPs) to be reported depending on the clinical site, adding a PRS for one clinical site and hiding it from others, displaying a pharmacogenomic section for some sites and modifying the content of that section depending on site preferences, and modifying which set of metadata was displayed depending on the clinical site. Neptune implemented these customizations by employing a templating system that can key off sample-specific metadata that is extracted from the LIMS.
Genomic variants were interpreted according to ACMG/AMP guidelines [37] externally from Neptune and stored in the VIP database, in a high-throughput manner that relied on a set of automated filters, defined prior to the project start. In general, manual review of variants is the exception. In eMERGE over 99.99% (682,343/682,398) from a representative sample) of variants were handled automatically, and in a recent batch we see recall of 100%, precision of 26.4%, f1 measure of 41% and specificity of 99.99% (Supplementary Tables 3,4). We employed a defined process for handling variant harmonization that has been previously described [28]. We started with a single reviewer who handled all variant interpretation and report sign-out activities. Later, we added a small team of 2–4 second reviewers and a dedicated first reviewer. Taking advantage of recurrent variant interpretations using the VIP database, we observed a rapid decline in novel variants per sample, followed by a stabilization around one reviewable variant per sample (Fig. 2). A key lesson learned was the benefit of gene-centric reviews; we adopted a review approach that “batched” together a large number of samples (typically 1,200), and then reviewers curated all variants in a particular gene from this batch in a single session. For example, a typical batch might contain 10 rare BRCA2 variants; these would all be interpreted in the same session by one reviewer. This approach reduced context switching for reviewers, streamlined literature review, and simplified adding additional members to the review team. The change proved to be popular with the review team and will be applied to future projects.
We engaged in multiple reanalysis activities as part of eMERGE III, supported by Neptune. First, we compared two snapshots of the ClinVar download (available from ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/), from August 2018 and August 2019. Variants with a new pathogenic or likely pathogenic (P/LP) interpretation where there were none previously were considered candidate upgrades. Variants where a previous P/LP assertion had been removed, leaving only variants of uncertain significance (VUS), benign, or likely benign, were candidates for a classification downgrade. In the genomic regions covered by our test, we identified 614 unique variants with changed assertions. For potential downgrades, we only considered variants that we had previously reported as P/LP, as many of the new ClinVar entries supported our decision during reporting to not report a variant that had been previously classified as P/LP in ClinVar. The result of this filtering was 109 unique variants to review (99 upgrades, 10 downgrades) of which 34 (28 upgrades, 6 downgrades) of these had 2, 3, or 4 stars in the August 2019 ClinVar snapshot (indicating multiple submitters with no conflicts, expert panel review, or practice guideline, respectively). For each of these variants, we performed a full, manual variant interpretation, considering all ACMG/AMP evidence categories. Ultimately, we found five variants with sufficient evidence to change the variant interpretation and issued corrected reports. The total time required for manual review varied greatly from between a few minutes and >5 hours, based primarily on the additional information available about the variant and the number of discussions required by the review team to finalize their interpretation. For first review, reanalysis took 32 minutes on average (SD 9.4). The majority of variants could be reclassified by a first reviewer, but a small fraction (<9%) required attention from a laboratory director.
In a separate reanalysis activity, we identified genomic VUS that, with the addition of one ACMG/AMP subcategory, could reach P/LP status. As the phenotypic and family history information gathered during eMERGE was quite limited, we requested a manual chart review from clinical sites for these variants (Fig. 3b). There were 83 variants identified initially, of which we reclassified 4, either using ACMG/AMP subcategory PS4 (prevalence in affecteds significantly increased over controls) or PP4 (patient’s phenotype or family history highly specific for gene). An example was the NM_000551.3:c.551T>C variant in the VHL gene, which was borderline VUS based on the evidence we had (PP3: computationally predicted to be deleterious, PM2: absent from population databases). Two papers reported the variant associated with affected individuals, but this was not enough evidence to apply PS4. However, upon contacting the clinical site, we learned that the patient was diagnosed with von Hippel–Lindau disease, which allowed us to apply the PP4 subcategory, moving this variant to LP status.

Neptune supported two parallel reanalysis activities during the eMERGE III project. The first was a project with the goal of providing updated reports when variant classifications change (a) over time. To accomplish this, we used Neptune’s reanalysis module to compare a ClinVar snapshot to local variant categorizations. We identified upgrades and downgrades by detecting either unreported variants with a new pathogenic/likely pathogenic (P/LP) classification in ClinVar or a reported variant with a new variant of uncertain significance (VUS), benign, or likely benign classification. There were 26 upgrades for review, resulting in 3 updated reports (all initially VUS) and 86 downgrades for review, resulting in 2 updated reports. Next, we collected a set of VUS variants that were lacking one American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) subcategory to reach an overall classification of LP (b). We then contacted clinical sites requesting more detailed patient phenotype information, to be able to apply the PP4 ACMG/AMP subcategory (patient phenotype or family history highly specific for gene). In four cases we were able to issue updated reports, all due to the new clinical information. In a separate study, we reanalyzed 83 variants based on additional clinical information requested from clinical sites for variants that were VUS but that could be reclassified as LP with the application of one ACMG subcategory. This resulted in four updated reports and highlights the importance of detailed clinical information during review by clinical geneticists.
In total, we reissued nine reports based on variant classification updates. By using the size of the eMERGE panel (68 consensus genes) and the number of reports in circulation when we started that effort (approximately 15,000) we can estimate that the burden placed on clinical laboratories by reanalysis will require assessing 0.0001 (109/1,020,000) variants per gene on an issued report. The rate of reissued reports remains low, at 0.03% (5/15,000). As the number of interpreted variants increases, this problem will continue to grow.
Case study: BCM HeartCare
In a second application, we performed variant interpretation and reporting for 709 patients who presented at BCM cardiovascular clinics. Of these cases, 8.5% were positive for a P or LP SNV or CNV, and 49% were positive for a pharmacogenomic finding. Management changes as a result of these findings included recommending additional specific laboratory testing including imaging, referral for a genetic consultation, or a change in medication.
For HeartCare, our review team of 2–4 analysts handled the initial variant reviewers, while a dedicated clinical geneticist with expertise in cardiovascular genetics handled the final review and report sign out. Discordances with groups outside of the project are handled by the reanalysis process. A new addition was patient and family management recommendations, written by a clinical geneticist. This section provides feedback to the ordering physician on managing a genetic finding, and when appropriate contains advice on additional testing, drug regimens to start or avoid, additional genetic counseling, and recommendations on cascade testing. Composing the physician guidance section added significant amounts of time to report preparation. These changes were implemented by creating a new report template to support the additional fields. Supplementary Figure 2 shows an example HeartCare report.
Neptune enabled the reporting of structured PRS data for HeartCare. We implemented a previously developed PRS for coronary artery disease [38], based on 50 SNPs. High-risk individuals have a 91% higher relative risk of hospitalization after 10 years than low-risk individuals. In HeartCare, after clinician feedback, we reported the top 5% of individuals in this distribution as the “high-risk” group (top 5% ≥4.5824), which is somewhat more stringent than the original study [38]. The assessment of the clinical utility of these scores are ongoing, and the creation of clinical data sets in which PRS data are integrated with EMR data, enabled by tools like Neptune, will aid these assessments.
We also implemented a HIPAA-compliant reporting portal, hosted on Amazon Web Services (AWS), for the final report rendering and storage. We piloted an integration of this reporting platform with Epic. This required generating HL7v2 messages that contain the encoded clinical report and key report results using the HAPI api (https://hapifhir.github.io/hapi-hl7v2/). The Epic team developed a new interface for displaying this information, and a new data model for storing it. HL7 messages were transferred by sftp, and automatically loaded by Epic and attached to the test order. To keep the HL7 message simple we included fields for the order number, Medical Record Number (MRN), test name, environment, last name, first name, middle initial, date of birth (DOB), gender, visit number, HGSC accession, observation date, specimen received date, ordering provider, results report date, result status, LP(a) finding, genetic finding, and address. Supplementary figure 3 shows an example of how this data appeared in Epic for ordering providers. In coordination with the Epic team, we tested the functionality, performance, and security of this approach using HL7 messages from 32 samples. These samples were loaded by the Epic team who then shared screenshots of the Epic interface and PDF reports for review. At the conclusion of the HeartCare project, we had successfully connected Neptune to Epic and ensured the resulting interface was secure, performant, and that data were received correctly by Epic. A full description and lessons learned from the HeartCare study are described in Murdock et al. 2021 (under review).
DISCUSSION
Neptune provides a customizable platform that enables the delivery of genomic results to support genomic medicine. It facilitates complex reporting workflows including reanalysis, and connects genomic data to clinical geneticists and the EHR. It is backed by a VIP database of genetic variation that stores variant curations. We have utilized this environment to enable two exemplar projects in which clinical genetic data were reviewed, reported out, and transferred back to a clinical site. Neptune is a validated approach to clinical genetic reporting that can alleviate some of the problems related to delivering scalable clinical genetic data.
Reanalysis places a substantial workload on clinical genetics activities and the overall effort will increase with the volume of reports issued. Based on the number of genes present on the gene panel designs used in the tests reviewed here, we observed a rate of 0.0001 variants per gene on an issued report per year. Thus, when reporting clinical genetic data at a large scale, complete reanalysis may not be feasible and clear guidelines will be crucial to define the extent to which reanalysis activities are necessary. Future work will examine the extent to which accelerating submissions to ClinVar might change this estimate and whether potential increasing concordance between laboratories will reduce the amount of work required.
The approach to variant review presented here relies on manual interpretation of variants, and thus has limitations to scalability as the number of reported genes increases to, e.g., an exome. This limit is evident in the plateau that is reached in the review burden per sample (Fig. 2) as additional samples are added to the study that we and others have observed [39]. Based on harmonization activities that we have conducted with other labs [28, 40] the approach here is consistent with best practices in the field, and scaling variant interpretation is likely to be a general challenge for the field in the coming years. Active efforts toward rule-based interpretation underway by ClinGen will help automatable genomic variant interpretation become standard.
The challenge of integrating genomic data into an EHR was made clear during HeartCare, where developers were unable to access the Epic test environment directly. Instead, our testing methodology relied on sharing screenshots for review, resulting in many slow iterations. Simplifying the HL7 message itself also proved to be key. A more complex message would have required still more rounds of testing and would have been challenging to review in multiple views in Epic. A surprising challenge was the difficulty of receiving confirmation from Epic for correct receipt of a message. This feature required additional configuration in Epic but was essential for the smooth operation of clinical reporting. Finally, we only started exploring the patient experience, but this aspect of the project is critical and should be a focus from the outset. True interoperability with the EMR will require the ability to extract de-identified data, which can be useful during variant interpretation and discovery. This level of interaction has not been achieved yet by our systems, but will be a future goal.
The successful implementation of genomic medicine relies on structured integration of genomic data into the EHR systems. These data cannot remain in silos; rather, they should be shared as widely as possible given the constraints of research consent and PHI data protection. When stored in a structured format, these data can be acted on by Clinical Decision Support (CDS) tools to provide context-dependent decision support to clinicians. Optimally, data would flow smoothly both into and out of the EHR. Health information can be used to support variant interpretation and genomic data are already proving actionable in the clinic, with its utility increasing rapidly. Data interchange formats like FHIR (https://emerge-fhir-spec.readthedocs.io/en/latest/) are crucial for enabling this interchange and will empower the next generation of clinical genomic integration.
Data availability
Data are available in dbGaP for controlled public access (phs001616.v1.p1).
The software is available from https://gitlab.com/bcm-hgsc/neptune.
References
Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature. 2015;526:336–42.
McPadden J, Durant TJ, Bunch DR, Coppi A, Price N, Rodgerson K, et al. Health care and precision medicine research: analysis of a scalable data science platform. J Med Internet Res. 2019;21:e13043.
Johnson A, Zeng J, Bailey AM, Holla V, Litzenburger B, Lara-Guerra H, et al. The right drugs at the right time for the right patient: the MD Anderson precision oncology decision support platform. Drug Discov Today. 2015;20:1433–8.
Alzu’bi A, Zhou L, Watzlaf V. Personal genomic information management and personalized medicine: challenges, current solutions, and roles of HIM professionals. Perspect Health Inf Manag. 2014;11:1c.
Erlich Y, Williams JB, Glazer D, Yocum K, Farahany N, Olson M, et al. Redefining genomic privacy: trust and empowerment. PLoS Biol. 2014;12:e1001983.
Holt JM, Wilk B, Birch CL, Brown DM, Gajapathy M, Moss AC, et al. VarSight: prioritizing clinically reported variants with binary classification algorithms. BMC Bioinformatics. 2019;20:496.
Rehm HL. Evolving health care through personal genomics. Nat Rev Genet. 2017;18:259–67.
Huang BE, Mulyasasmita W, Rajagopal G. The path from big data to precision medicine. Expert Rev Precis Med Drug Dev. 2016;1:129–43.
Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3:16.
Manolio TA, Chisholm RL, Ozenberger B, Roden DM, Williams MS, Wilson R, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15:258–67.
Vozikis A, Cooper DN, Mitropoulou C, Kambouris ME, Brand A, Dolzan V, et al. Test pricing and reimbursement in genomic medicine: towards a generalstrategy. Public Health Genomics. 2016;19:352–63.
Consortium TE, The eMERGE Consortium, Gibbs RA, Rehm HL. Harmonizing clinical sequencing and interpretation for the Emerge III Network. Am J Hum Genet. 2019;105:588–605.
IAll of Us Research Program Investigators, et al. The “All of Us” Research Program. N Engl J Med. 2019;381:668–76.
Weitzel KW, et al. The IGNITE network: a model for genomic medicine implementation and research. BMC Med Genomics. 2016;9:1.
Amendola LM, Berg JS, Horowitz CR, Angelo F, Bensen JT, Biesecker BB, et al. The Clinical Sequencing Evidence-Generating Research Consortium: integrating genomic sequencing in diverse and medically underserved populations. Am J Hum Genet. 2018;103:319–27.
Williams MS. Early lessons from the implementation of genomic medicine programs. Annu Rev Genomics Hum Genet. 2019;20:389–411.
Dewey FE, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814.
eMERGE. Results FHIR specification—emerge-fhir-spec 1.0 documentation, 2021. https://emerge-fhir-spec.readthedocs.io/en/latest/. Accessed 28 April 2021.
AI Genome Analysis & Reporting Platform, 2021. https://fabricgenomics.com/. Accessed 28 April 2021.
SOPHiA Genetics. https://www.sophiagenetics.com/en_US/home.html. Accessed 28 April 2021.
Machine learning genomic analysis platform. https://www.emedgene.com/. Accessed 28 April 2021.
PharmCAT. http://pharmcat.org/. Accessed 28 April 2021.
Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6.
Lassmann T, Francis RW, Weeks A, Tang D, Jamieson SE, Broley S, et al. A flexible computational pipeline for research analyses of unsolved clinical exome cases. NPJ Genom Med. 2020;5:54.
Hussain MA, Langer SG, Kohli M. Learning HL7 FHIR using the HAPI FHIR server and its use in medical imaging with the SIIM dataset. J Digit Imaging. 2018;31:334–40.
Shirts BH, Salama JS, Aronson SJ, Chung WK, Gray SW, Hindorff LA, et al. CSER and eMERGE: current and potential state of the display of genetic information in the electronic health record. J Am Med Inform Assoc. 2015;22:1231–42.
Reid JG, Carroll A, Veeraraghavan N, Dahdouli M, Sundquist A, English A, et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics. 2014;15:30.
eMERGE Consortium. Harmonizing clinical sequencing and interpretation for the eMERGE III Network. Am J Hum Genet. 2019;105:588–605.
Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7 -associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20:351–9.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Chiang T, Liu X, Wu T-J, Hu J, Sedlazeck FJ, White S, et al. Atlas-CNV: a validated approach to call single-exon CNVs in the eMERGESeq gene panel. Genet Med. 2019;21:2135–44.
Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19:581–90.
Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89:464–467.
Aronson S, Babb L, Ames D, Gibbs RA, Venner E, Connelly JJ, et al. Empowering genomic medicine by establishing critical sequencing result data flows: the eMERGE example. J Am Med Inform Assoc. 2018;25:1375–81.
Senol-Cosar O, Schmidt RJ, Qian E, Hoskinson D, Mason-Suares H, Funke B, et al. Considerations for clinical curation, classification, and reporting of low-penetrance and low effect size variants associated with disease risk. Genet Med. 2019;21:2765–73.
Schwartz GG, Ballantyne CM, Barter PJ, Kallend D, Leiter LA, Leitersdorf E, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol. 2018;3:164–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–58.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Amendola LM, Muenzen K, Biesecker LG, Bowling KM, Cooper GM, Dorschner MO, et al. Variant classification concordance using the ACMG-AMP variant interpretation buidelines across nine genomic implementation research studies. Am J Hum Genet. 2020;107:932–41.
Acknowledgements
This work was funded by internal operating funds of the Baylor College of Medicine Human Genome Sequencing Center (HGSC), and by the National Institutes of Health (NIH) eMERGE program Phase III: U01HG8657 (Kaiser Permanente Washington/University of Washington); U01HG8685 (Brigham and Women’s Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children’s Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children’s Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine).
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: E.V., R.G. Data curation: A.S., S.L., Q.M., X.T., M.C., D.M. Formal analysis: Funding acquisition: R.G, D.M. Investigation: E.V., M.C., D.M. Methodology: E.V., R.G., V.Y., C.K. Project administration: M.C., C.K., M.M. Resources: W.W., W.C., C.W., G.W., G.J., R.G. Software: E.V., V.Y., S.K., T.W., Supervision: E.V., M.M., D.M., C.K. Validation: E.V., V.Y., T.W. Visualization: E.V., M.C. Writing—original draft: E.V., V.Y. Writing—review & editing: E.V., V.Y., D.M., S.K., T.W., A.S., S.L., Q.M., X.T., M.M., M.C., C.K., W.W. W.C., C.W.,G.W., G.J., D.M., R.G.
Corresponding author
Ethics declarations
Ethics Declaration
The Electronic Medical Records and Genomics (eMERGE) Network is a National Human Genome Research Institute (NHGRI)-funded consortium tasked with developing methods and best practices for utilization of the electronic medical record (EMR) as a tool for genomic research. All 11 sample collection sites consented participants under institutional review board (IRB)-approved protocols and the two sequencing centers had IRB-approved protocols that deferred consent to the participating sites. The protocol number for Baylor College of Medicine was(#H-40455).
Competing interests
E.V. is a cofounder of Codified Genomics, which provides variant interpretation services. R.G., D.M., D.M., disclose that the Baylor Genetics Laboratory is co-owned by Baylor College of Medicine. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Eric, V., Yi, V., Murdock, D. et al. Neptune: an environment for the delivery of genomic medicine. Genet Med 23, 1838–1846 (2021). https://doi.org/10.1038/s41436-021-01230-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01230-w
This article is cited by
-
The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities
Communications Biology (2024)
