
Abstract
Purpose
Phosphatidylinositol Glycan Anchor Biosynthesis, class G (PIGG) is an ethanolamine phosphate transferase catalyzing the modification of glycosylphosphatidylinositol (GPI). GPI serves as an anchor on the cell membrane for surface proteins called GPI-anchored proteins (GPI-APs). Pathogenic variants in genes involved in the biosynthesis of GPI cause inherited GPI deficiency (IGD), which still needs to be further characterized.
Methods
We describe 22 individuals from 19 unrelated families with biallelic variants in PIGG. We analyzed GPI-AP surface levels on granulocytes and fibroblasts for three and two individuals, respectively. We demonstrated enzymatic activity defects for PIGG variants in vitro in a PIGG/PIGO double knockout system.
Results
Phenotypic analysis of reported individuals reveals shared PIGG deficiency–associated features. All tested GPI-APs were unchanged on granulocytes whereas CD73 level in fibroblasts was decreased. In addition to classic IGD symptoms such as hypotonia, intellectual disability/developmental delay (ID/DD), and seizures, individuals with PIGG variants of null or severely decreased activity showed cerebellar atrophy, various neurological manifestations, and mitochondrial dysfunction, a feature increasingly recognized in IGDs. Individuals with mildly decreased activity showed autism spectrum disorder.
Conclusion
This in vitro system is a useful method to validate the pathogenicity of variants in PIGG and to study PIGG physiological functions.
Similar content being viewed by others

INTRODUCTION
Phosphatidylinositol Glycan Anchor Biosynthesis, class G (PIGG) is an ethanolamine phosphate transferase that catalyzes the modification of the second mannose of glycosylphosphatidylinositol (GPI). It is 1 of the 22 phosphatidylinositol glycan (PIG) genes involved in the biosynthesis of GPI.1 GPI serves as an anchor for over 150 surface proteins on cell membrane. These proteins, called GPI-anchored proteins (GPI-APs), play a variety of essential roles throughout the human body but are especially critical for development and neurogenesis.2 GPI and GPI-AP are linked together by a bridge of ethanolamine phosphate (EtNP) on the third mannose catalyzed by PIGO.3 Variants in genes involved in the biosynthesis of GPI are known to cause inherited GPI deficiency (IGD). Typically, IGDs cause a decrease of GPI-AP levels on blood cell surface.2 They are also associated with hyper/hypophosphatasia because the lack of, or abnormal, GPI anchor on alkaline phosphatase triggers its secretion out of the cell or its degradation.3,4,5 Currently 21 genes are associated with IGDs.6 Their main clinical characteristics are intellectual disability, seizures, and facial dysmorphisms.7
Previously, three studies have described seven individuals from five unrelated families with biallelic PIGG variants.1,3,8 Surprisingly, two of these studies described normal surface levels and normal structure of GPI-APs on granulocytes—despite an almost complete inactivation of PIGG.
Herein, we report 19 unrelated families with biallelic PIGG variants. We describe characteristic features of this entity and introduce a functional analysis to validate pathogenicity of PIGG variants.
MATERIALS AND METHODS
Identification of affected individuals and clinical information collection
Subjects were recruited via the Deciphering Developmental Disorders (DDD) study,9 GeneMatcher,10 or by an international network of collaborating clinicians.
Analysis of PIGG variants
PIGG variants were compared in Varsome.11 Mean and highest minor allele frequency from gnomAD were extracted. Pathogenicity scores from the American College of Medical Genetics and Genomics (ACMG) and dbNSFP were compiled. CADD scores were obtained from https://cadd.gs.washington.edu.12 PIGG variants and its domains from UniProt13 were shown in ProteinPaint14 and in Protter.15 Alignments for residue conservation was done using UCSC Genome Browser.16
Blood fluorescence-activated cell sorting
Analyses on granulocytes were performed on one blood sample per individual in two technical replicates. Granulocytes were stained on ice with markers for 1 hour. Markers used were phycoerythrin (PE)-conjugated antihuman CD16 (BioLegend), fluorescein isothiocyanate (FITC)-conjugated mouse antihuman CD55 and CD59 (BD PharMingen), and FLAER-Alexa448 (Cedarlane). FACS Lysing Solution (BD Bioscience) was used to lyse red blood cells. Fibroblasts could be obtained for two individuals. Fibroblasts were cultivated in DMEM, harvested at 80–90% confluency, and stained with markers in an incubation buffer containing 0.5% bovine serum albumin (BSA) for 1 hour on ice. Markers used were FLAER-Alexa448, FITC-conjugated mouse antihuman CD73 (Biolegend), and PE-conjugated mouse antihuman CD109 (BioLegend). Analysis was done on three independent experiments with FlowJo software after samples were washed using a BD FACSCanto II system (BD Biosciences).
Functional analysis of PIGG variants
PIGG/PIGO double knockout (DKO) cells were generated from HEK293 cells using the CRISPR/Cas System. Single PIGOKO and PIGGKO cells were reported in previous papers.3,17 DKO cells were generated by further knockout of PIGG on single PIGOKO cells. Guide RNA used for PIGGKO is in exon 1 (gcgtagcgatcgaggtgcta) and exon 2 (gccctacacaacttaccttg). The DKO clones were obtained by limiting dilution and further selected by fluorescence-activated cell sorting (FACS) analysis, polymerase chain reaction (PCR), and direct Sanger sequencing (Fig. S1). PIGOKO cells show partial loss of surface GPI-APs, which is completely removed by further knockout of PIGG. One clone was chosen for functional assays. Wild-type and variant PIGG are then used to rescue GPI-AP expression on PIGO/PIGG DKO cells, which can measure the PIGG variant activity. DKO cells were transiently transfected with a wild-type or mutant PIGG complementary DNA (cDNA) cloned into a strong promoter-driven (SRα) expression vector, pME PIGG-GST or a weak thymidine kinase promoter-driven expression vector, pTK PIGG-GST. To determine transfection efficiency, luciferase expression plasmid was co-transfected with PIGG-GST plasmids. Restoration of the surface expression of DAF was analyzed two days later by staining cells with anti-DAF antibody (clone IA10) followed by PE labeled antimouse IgG and analyzed by flow cytometry. PIGG variant activity was shown by the percentage of geometric mean of DAF fluorescent intensity of each PIGG variant transfectant against that of the PIGG wild transfectant. The protein expression of each PIGG-GST variant was analyzed by western blotting with anti-GST antibody (antigoat GST, GE Healthcare) using the cell lysate of each transfectant. Luciferase activities in the same cell lysates were also determined. To determine PIGG-GST levels, band intensities of PIGG-GST were divided by the band intensities of GAPDH (loading control) and further by luciferase activities (transfection efficiency). Levels are shown in arbitrary units. Experiments were repeated at least twice.
RESULTS
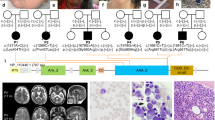
Nineteen unrelated families were recruited for this study, including 22 individuals with 22 different PIGG variants. Family number was given according to enzymatic activity reduction of variants (null activity to no decrease). Individual 8A was reported previously by Lionel et al. but without detailed clinical description.8 Consanguinity was present in nine families (Fig. 1). Individuals with biallelic PIGG variants all presented developmental delay/intellectual disability (DD/ID) except for individual 15. Individual 15 did not show DD but is too young to be evaluated for ID. A subjective severity score was given to each characteristic based on clinical information provided by clinicians (Table 1, blue color gradient shows degrees of enzymatic activity reduction). DDs were mixed, affecting motor, language, and social aspects. Seizures affected 13 individuals in this cohort (62%). For all affected individuals, seizures were mostly febrile and occurred mainly in the first two years of life. Almost all individuals are now seizure free (except individuals 4 and 19). Electroencephalogram (EEG) showed mostly diffuse slowing and generalized spikes. Twelve individuals presented hypotonia (57%). Hypotonia was particularly severe in individuals 4, 8A, and 10 and was generalized. Ataxia and nystagmus were also prominent features in this cohort, affecting 9 (43%) and 8 (38%) individuals, respectively. Individual 5B demonstrated a familial hemiplegic migraine/episodic ataxia phenotype. Hemiplegic migraines were well-controlled with amitriptyline, but she was wheelchair-bound because of frequent falls, painful myokymia, and wide-based gait. Individual 18 had acute onset ataxic attacks with regression around 2 years old that responded to high dose corticosteroids. Strabismus and tremor were also observed in five individuals each (24%). Tremors were intentional and/or resting. Individual 13B had esotropia in infancy that resolved. Five individuals had diminished deep tendon reflexes (24%). In individual 4, reflexes were normal in the upper limbs but diminished in the lower limbs, unlike individual 9 who had brisk reflexes. Minor dysmorphisms were observed in four individuals (19%). They variably had smooth and long philtrum, thin upper lip, shallow nose tip, tented mouth, coarse face, and mild bilateral fifth finger clinodactyly (Fig. 2a). Three individuals had microcephaly under the 2nd percentile (14%). Individual 17, however, had macrocephaly (head circumference in the 99th percentile). Autism spectrum disorders were diagnosed in two individuals and suspected in individual 16 who had autistic features (14%). Three individuals had short stature under the 10th percentile (14%). Magnetic resonance image (MRI) demonstrated cerebellar atrophy in eight individuals (38%) and normal alkaline phosphatase was found in all tested individuals (9; 43%). Common MRI findings included cerebellar, predominantly in the superior vermis, hypoplasia/atrophy that was progressive for individuals 2, 11, and 12 (Fig. 2b). Individual 19 was included separately in the figures and tables because we were unable to demonstrate that the phenotype was due to PIGG biallelic variants (see “Discussion“). Hearing loss and congenital heart defect were not common findings in this cohort, contrary to other IGDs. Individual 2 had a small spontaneously resolved atrial septal defect. Individual 7 showed mild unilateral hearing loss and small ventricular septal defect. Globally, most individuals seemed to clinically improve with age, with respect to DD, seizures, and ataxic features. Many affected individuals had extensive biochemical and genetic investigations with normal results (Table S2). Individual 13B had 6% absence of heterozygosity in her genome indicating consanguinity of the parents. Individual 14 was found to have a maternally inherited variant in the ELN gene that was predicted to be pathogenic. He had hyperelastic skin with easy bruising, bilateral congenital hip dysplasia, and joint hypermobility with Beighton score at 8/9. However, his electrocardiogram (ECG) and echocardiogram were normal and the mother had a normal skin and joint exam. We therefore considered this variant to be a variant of uncertain significance and included this individual in the study. Individual 5A had a skeletal muscle biopsy. Light microscopy and histopathology were normal. Electron microscopy showed a marked accumulation of subsarcolemmal mitochondria, some of which were enlarged and many of which had concentric cristae (data not shown). Spectrophotometry showed significant decrease in both complexes I and II. Mitochondrial DNA sequencing and metabolites were normal. Electron microscopy studies were normal in fibroblasts. Individual 8A also had a muscle biopsy at the age of three years showing subsarcolemmal aggregates of normal mitochondria on histopathology and electron microscopy and moderately low respiratory chain complex II enzymatic activity. Mitochondrial complex II deficiency was confirmed in skin fibroblasts. Other mitochondrial complex activities were normal in skin fibroblasts. This individual had decreased muscle bulk and weakness in the four limbs.

Filled squares correspond to affected males and filled circles correspond to affected females. Open squares or circles with a center dot correspond to male or female unaffected carriers.

(a) Photographs from individual 1 showing facial dysmorphism. This individual present with mildly coarse face, strabismus, long philtrum, and shallow nose tip. (b) Magnetic resonance image (MRI) from affected individuals showing cerebellar atrophy. Individual 8A: T1 weighted sagittal image shows minimal increase in the interfoliate distance of cerebellum superiorly at the age of 3 years. Individual 8B: T1 weighted sagittal image shows atrophy of the anterior lobe of the cerebellum (superior vermian atrophy) at the age of 2 years. Individual 13B: sagittal, coronal, and transverse MRI images at 12 years old showing cerebellar vermian atrophy predominantly affecting the superior vermis. Cerebellar atrophy was found in individuals 2, 3, 8A, 8B, 11, 12, 13A, 13B, and 19.
Individual from family 1 had a sibling with concordant phenotype but he was excluded from this study because he was not tested for PIGG variants (Fig. 1). We had no clinical details concerning this sibling. Consanguinity increases the risk for another recessive disease. Individual from family 6 had siblings with proven biallelic PIGG variants and seizures but clinical information was insufficient to include them in the study.
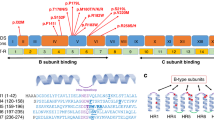
PIGG variants were dispersed throughout the protein length affecting amino acids positions 46 to 957 (See Fig. 3a, b). There was a higher density of variants from amino acid positions 115 to 138 around the metal-binding domain and positions 270 to 344 in the GPI ethanolamine phosphate transferase 2 domain near the second active site. Variants were 11 missenses, 10 nonsenses, and 1 splice variant. Variants p.Arg304Ter, p.Trp505Ter, p.Gln851Pro, and p.Leu875Ter were already reported in ClinVar as likely pathogenic or pathogenic. Missense variants were mostly located in luminal loops but two (p.Val531Met and p.Gln851Pro) were cytoplasmic. Variants p.Tyr934Ter and p.Tyr957Ter were the only two nonsense variants that were predicted to escape nonsense-mediated decay and to result in a truncated protein because of their position in the last exon. Amino acids 934 to 961 were strongly conserved across vertebrates, suggesting they may play an important role in PIGG physiology and that a nonsense variant in this region may have a deleterious impact (Fig. 3c). Individuals 11 and 12, who were respectively homozygous for p.Tyr934Ter and p.Tyr957Ter, showed incapacitating ataxia and progressive cerebellar atrophy. Seven residues affected by missense variants appeared to be strongly conserved, whereas four were poorly conserved (Met344, Ser497, Val531, Glu696). These four nonconserved residue variants were all found as compound heterozygous (individuals 18, 19, 16, 17, respectively). Individual 19 had a severe phenotype with respect to DD/ID and epilepsy. Individual 18 had severe acute ataxia and global DD. Individuals 16 and 17 had global DD, moderate ID, and severe language delay.

(a) Figure generated using ProteinPaint. Variants from affected individuals are in blue and matched according to each family’s genotype (F01 to F19). Transmembrane and luminal domains are from UniProt. Already published pathogenic variants are in green.1,3,8 ClinVar likely pathogenic and pathogenic variants are in yellow and red respectively. (b) Figure generated using Protter. Transmembrane domains are from UniProt and numbered 1 to 13. Missense variants are in pink and the two nonsense variants resulting in a truncated protein are in blue (p.Tyr934Ter and p.Tyr957Ter). (c) Conservation of affected residues and C-term region across vertebrates. Multiple alignment from UCSC Genome Browser.
We analyzed GPI-AP surface levels on granulocytes for individuals 8A, 8B, and 13A. FLAER, CD16, CD55, and CD59 were all normal in all individuals (Fig. 4a and S2). We analyzed fibroblasts from individuals 16 and 17. FLAER and CD73 were lower in both individuals compared to controls whereas CD109 was unchanged (Fig. 4b and S2).

(a) Fluorescence-activated cell sorting (FACS) from granulocytes of affected individuals. GPI-anchored proteins (GPI-AP) surface levels (FLAER, CD16, CD55, and CD59) on granulocytes for individuals 8A (red), 8B (green), and 13A (green) compared to their unaffected heterozygous mother (purple), father (blue), and unrelated controls (black). (b) FACS from fibroblasts of affected individuals. GPI-AP surface levels (FLAER, CD73, and CD109) on fibroblasts from individuals 16 and 17 (green) compared to control (black). (c) Functional assay using strong promoter-driven PIGG variants. HEK293 PIGO/PIGG DKO cells were transfected with strong promoter-driven (pME) wild-type or mutant PIGG complementary DNA (cDNA). Two days later, expression of DAF was analyzed by FACS. Most of the variants could not rescue the expression at all except F13, showing partial rescue. Blue percentages indicates % of rescue compared to wild-type PIGG. PIGG variant activity was shown by the percentage of geometric mean of DAF fluorescent intensity of each PIGG variant transfectant against that of the PIGG wild-type transfectant. (d) Functional assay using the weak promoter to increase sensitivity for variant dysfunction. HEK293 PIGO/PIGG DKO cells were transfected with strong promoter-driven (pME) or a weak promoter-driven (pTK) wild-type or mutant PIGG cDNA. Two days later, expression of DAF was analyzed by FACS except for F17 (pTK), showing expression of CD59. All variants could rescue similar level to wild-type PIGG when using strong promoter (100% of rescue). Driven by weak promoter, variants showed various level of restoration of GPI-APs. PIGG variant activity was shown by the percentage of geometric mean of DAF fluorescent intensity of each PIGG variant transfectant against that of the PIGG wild-type transfectant.
Eighteen different variants were tested in the PIGO/PIGG DKO system. Restoration of the GPI-AP expression on PIGO/PIGG DKO cells by transfection with wild-type or variant PIGG cDNA was compared (Fig. 4c, d). PIGG variants’ expression was analyzed by western blotting (Fig. S3). Variants p.Ala46GlnfsTer28, p.Thr115TyrfsTer50, and p.Phe580LeufsTer2 were not tested but their activities were expected to be null, as they were frameshift variants. In contrast, variant p.Trp505Ter was expected to have decreased activity because it was similar to previously published p.Trp547Ter variant.1 This variant was shown to result in moderate and mild phenotype because of residual full-length protein from read through. From our functional assay, ten variants had a null enzymatic activity, one had severe decrease in activity, two had a decreased activity, four had a mild decrease in activity, and two had slight or no decreases in activity. The prediction tools could clearly differentiate pathogenic variants with null or decreased activity from benign variants with no or slight decreased activity, but they could not predict the pathogenicity of the variants with mild decreased activity (Table S1). The variants with null, severely decreased, and decreased activity variants had a mean pathogenicity CADD score of 29.5. Variants causing slight or no decrease were predicted to be likely benign variants and had a mean CADD score of 11.7. Both variants of individuals 11 and 12 (p.Tyr934Ter and p.Tyr957Ter) truncated the strongly conserved C-terminal end of PIGG resulting in a null enzymatic activity (Fig. 3c). Prediction tools predicted these variants to be likely pathogenic and CADD scores were 31 and 36, respectively. Individuals 16, 17, and 18 had one of their variants located in a nonconserved residue (Met344, Val531, and Glu696), with slight/no or mild decrease and CADD scores of 0.61, 9.0 and 6.0 respectively. All individuals with PIGG deficiency except one showed DD/ID but the severity did not correlate with the degree of decreased activities (Table 1). Individuals 8A and 9 showed severe DD/ID, whereas individuals 3, 4, 5B, 8B, 11, and 12 showed mild DD/ID, although their PIGG variants were all with null activity. On the contrary, individual 17, the two variants of which had mildly decreased activity showed moderate DD/ID. One variant (p.Arg681Trp) had a pathogenicity CADD score of 32. Affected individuals with null to severely decreased activity showed higher probability of cerebellar atrophy (individuals 2, 3, 8A, 8B, 11, 12, 13A, and 13B), seizures, tremor, nystagmus, diminished tendon reflexes, strabismus, dysmorphism, microcephaly, and short stature (Fig. S4). The most severely affected individuals regarding DD/ID (individuals 8A and 9) and hypotonia (Individuals 4, 8A, and 10) were homozygous for null activity variants. Individuals with milder activity decrease showed statistically significant higher probability of autistic features (individuals 14, 16, 17). Overall, individuals with null or severely decreased enzymatic activity showed a statistically higher number of phenotypic features than individuals with decreased, mildly decreased, or slightly decreased enzymatic activity (Fig. S5).
DISCUSSION
Our study of 22 individuals with PIGG deficiency further delineates and confirms the phenotypic description previously reported in smaller studies.1,3,8 PIGG is involved in GPI biosynthesis6 and is widely expressed.18,19 However, individuals with PIGG deficiency mainly showed neurological abnormalities, suggesting its important role in neurological development.1,3,8 Individuals with biallelic pathogenic variants in PIGG showed DD/ID, hypotonia, early-onset seizures, cerebellar atrophy, ataxia, and minor facial dysmorphisms. We here report several additional PIGG deficiency–associated features: nystagmus, strabismus, tremor, diminished deep tendon reflexes, microcephaly, autism spectrum disorder, and short stature, which were found in 14% or more of individuals from this cohort. Strabismus had been reported in one individual in Zhao et al.1 Hyporeflexia, autistic features, and growth retardation were also described in Makrythanasis et al.3 but were not present in a significant proportion of individuals. Contrary to what is seen in several other IGDs, individuals with PIGG deficiency have normal levels of alkaline phosphatase and often normal levels of GPI-APs on granulocytes. DD/ID, seizures, hypotonia, and facial dysmorphisms, which are classic IGD phenotypic features, are also common with PIGG deficiency symptoms. Other characteristics of IGDs, such as anorectal and finger anomalies, hearing loss, and kidney hypoplasia,3 were not found in this cohort of individuals with biallelic PIGG variants.
As for the genotype–phenotype correlation, affected individuals with PIGG variants of null or severely decreased activity showed a trend toward tremor, cerebellar abnormalities, diminished deep tendon reflexes, strabismus, dysmorphism, microcephaly, short stature, and mitochondrial dysfunction. They also presented with a significantly higher number of phenotypic features than other affected individuals. On the other side of spectrum, those with mild decrease showed a statistically significant association with autism spectrum disorder. Some features like DD/ID and hypotonia were more ubiquitous throughout individuals with biallelic PIGG variants regardless of their variant’s enzymatic activity. The most severely affected individuals regarding these two features had homozygous null variants but some individuals with homozygous null variants had a milder phenotype. Seizure was also a common symptom regardless of enzymatic activity, which might also affect DD/ID severity. Individuals 8A, 9, and 10, who shared the same genotype (homozygous p.Leu875Ter), were similar in the severity of their DD/ID. In addition, although the siblings from families 5, 8, and 13 broadly resembled each other in terms of their intrafamilial neurological manifestations, clinical severity was different between siblings. Siblings 8A and 8B, despite the similarities of their brain MRI findings, surprisingly had dissimilar degrees of ID and hypotonia. This difference could potentially be explained by the presence of an additional mitochondrial complex deficiency in individual 8A. We initially expected that clinical severity would correlate with the reduction of enzyme activity, but the affected individuals have different genetic backgrounds and environments. This might explain why strict correlation cannot be obtained.
Individual 19 had a very severe phenotype, especially regarding DD/ID, seizures, strabismus, and cortical blindness. Our functional assay, however, did not show any decrease for the Ser497Leu variant. Pathogenic predictions were in favor of a likely benign variant and the residue’s conservation in vertebrates was incomplete. Moreover, the second variant for this individual, p.Thr124Met, only resulted in a mild activity decrease but the conservation for this residue was high and pathogenic predictions were unclear. It is possible that even a mild decrease in enzymatic activity, too mild to be detected by our assays, could affect neurons and have clinical repercussions. There were no alternative likely causative variants found on genome sequencing including sequence and structural variants as well as mitochondrial variants. Therefore, at this point, we and the caregivers are uncertain as to the involvement of these PIGG variants in the phenotype of this individual. Thus, because of the phenotype severity combined with these multiple evidence of low pathogenicity for both variants, we decided to exclude individual 19 from our phenotype analysis and her clinical description was separated in Table 1 and Table S2.
It is puzzling that several individuals with IGDs have features of mitochondrial disease. Here, we report two individuals with a complex II deficiency in addition to a PIGG deficiency (individuals 5A and 8A). Individual 5A also had complex I deficiency. Individual 8A was reported to have mitochondrial complex II deficiency in a previous study.8 Tarailo-Graovac et al. described a case of PIGA deficiency with mitochondrial complex deficiency and suggested that several mitochondrial membrane proteins may be associated with GPI-APs, or GPI-anchored themselves.20,21 There is a need to investigate the mechanism and the strength of the association of mitochondrial dysfunction with different GPI deficiency disorders. In the meantime, it could be reasonable to evaluate mitochondrial function in individuals with IGD and to exclude IGDs in individuals with unexplained mitochondrial phenotype.
In individuals with IGD and involvement of other enzymes in the GPI biosynthesis pathway, the most reduced GPI-APs on flow cytometry are CD16 in granulocytes and CD73, as well as CD109 in fibroblasts.2,22,23 In contrast, the GPI-AP studies of PIGG-deficient granulocytes are not typical of those usually seen in other IGDs. There is often no reduction in GPI-AP levels nor changes in their structure on granulocytes.3 Our results are consistent with previous studies as we did not find changes of GPI-AP levels on granulocytes. Indeed, fibroblasts seem to be the best tissue for PIGG deficiency diagnosis—to look both at GPI-AP levels and at the consequences on mitochondrial function.24,25 Zhao et al. found that GPI-APs are decreased on PIGG-deficient fibroblasts and CD73 was the lowest marker compared to controls.1 Similarly, in our study, CD73 was also the most reduced marker in both tested individuals. GPI-AP levels diminution on fibroblasts corroborate our findings that PIGG variants from individuals 16 and 17 are pathogenic. Contrarily to other IGDs, CD109 was normal in the two affected individuals. It has been well established that proteins are attached to EtNP linked to the third mannose.6 PIGG is involved in transfer of EtNP to the second mannose before attachment of proteins. In normal cells, the PIGG-dependent EtNP is present in the nascent GPI-APs and is then shortly removed from GPI-APs.6 In PIGG-defective cells, the protein attachment occurs to GPI lacking EtNP on the second mannose. Apparently, this situation did not appreciably affect GPI-anchoring of many proteins in granulocytes, however, the same situation significantly affected GPI-anchoring of CD73, but not CD109, in fibroblasts. Similarly, alkaline phosphatase level was not affected as it was normal in all tested individuals. Further study is necessary to clarify mechanistic basis of these variable requirements of PIGG in different cell types and for different GPI-APs.
In this study and previously published work, cerebellar atrophy is a common feature among individuals with PIGG deficiency (12/28), as it is in other IGDs, such as PIGH, PIGO, and GPAA1.5,23 Alkaline phosphatase, which has a GPI-anchored form, is important to get vitamin B6 in neurons to help GABA synthesis. Some published individuals with other GPI biosynthesis defects have B6-responsive seizures.5,22,26,27 For IGDs in general, we recommend measuring serum alkaline phosphatase and supplementing pyridoxine and/or pyridoxal phosphate. In individuals with PIGG deficiency, alkaline phosphatase is not useful in the diagnosis, but the supplements could potentially be tried for seizures. For new PIGG variant, a GPI-AP study on fibroblasts is ideal but not always possible because of the availability of the tissues. Also, the degree of GPI-AP decrease may not always reflect the level of pathogenicity of the variant. Given the lack of efficient method, we propose that our in vitro system could be a useful tool for diagnosis and research to validate pathogenicity of PIGG variants.
In conclusion, this study significantly enhances our understanding of the phenotypic range of individuals with biallelic pathogenic variants in PIGG. Not only DD/ID, hypotonia, seizures, cerebellar atrophy, ataxia and facial dysmorphism, but also other features, such as nystagmus, strabismus, tremor, diminished deep tendon reflexes, microcephaly, autism spectrum, short stature, and mitochondrial dysfunction have emerged as cardinal clinical signs of this disorder. Our study triples the number of reported individuals with PIGG deficiency. In addition, we propose an in vitro system for assessment of PIGG variant pathogenicity.
Data availability statement
Data and materials developed in support of the paper are available upon request.
References
Zhao, J. J. et al. Reduced cell surface levels of GPI-linked markers in a new case with PIGG loss of function. Hum. Mutat. 38, 1394–1401 (2017).
Murakami, Y. et al. Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in severe cases. Am. J. Hum. Genet. 105, 384–394 (2019).
Makrythanasis, P. et al. Pathogenic variants in PIGG cause intellectual disability with seizures and hypotonia. Am. J. Hum. Genet. 98, 615–626 (2016).
Murakami, Y. et al. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 287, 6318–6325 (2012).
Bellai-Dussault, K. et al. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 95, 112–121 (2019).
Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 10, 190290 (2020).
Bayat, A. et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia. 61, 1142–1155 (2020).
Lionel, A. C. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 20, 435–443 (2018).
Wright, C. F. et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 385, 1305–1314 (2015).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Kopanos, C. et al. VarSome: the human genomic variant search engine. Bioinformatics. 35, 1978–1980 (2019).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. (2019).
Zhou, X. et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 48, 4–6 (2016).
Omasits, U., Ahrens, C. H., Müller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 30, 884–886 (2014).
Lee, C. M. et al. UCSC Genome Browser enters 20th year. Nucleic Acids Res. 48, D756–D761 (2020).
Tanigawa, J. et al. Phenotype-genotype correlations of PIGO deficiency with variable phenotypes from infantile lethality to mild learning difficulties. Hum. Mutat. 38, 805–815 (2017).
Wu, C. et al. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 44, D313–6 (2016).
Su, A. I. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 (2004).
Zhao, P. et al. Proteomic identification of glycosylphosphatidylinositol anchor-dependent membrane proteins elevated in breast carcinoma. J. Biol. Chem. 287, 25230–25240 (2012).
Tarailo-Graovac, M. et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J. Rare Dis. 10, 23 (2015).
Nguyen, T. T. M. et al. Mutations in PIGS, encoding a GPI transamidase, cause a neurological syndrome ranging from fetal akinesia to epileptic encephalopathy. Am. J. Hum. Genet. 103, 602–611 (2018).
Nguyen, T. T. M. et al. Biallelic variants in the GPI transamidase subunit PIGK cause a neurodevelopmental syndrome with hypotonia, cerebellar atrophy, and epilepsy. Am. J. Hum. Genet. 106, 484–495 (2020).
Rodenburg, R. J. T. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 34, 283–292 (2011).
van den Heuvel, L. P., Smeitink, J. A. & Rodenburg, R. J. T. Biochemical examination of fibroblasts in the diagnosis and research of oxidative phosphorylation (OXPHOS) defects. Mitochondrion 4, 395–401 (2004).
Thompson, M. D. et al. Hyperphosphatasia with neurologic deficit: a pyridoxine-responsive seizure disorder? Pediatr. Neurol. 34, 303–307 (2006).
Kuki, I. et al. Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology 81, 1467–1469 (2013).
Acknowledgements
This work is supported by Canadian Institutes of Health Research (CIHR) and Fonds de Recherche du Québec–Santé (FRQS) awards to P.M.C. We thank Keiko Kinoshita, Saori Umeshita, Kae Imanishi (Osaka University) for technical help. T.S.B. is supported by the Netherlands Organisation for Scientific Research (ZonMW Veni, grant 91617021), a NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation, an Erasmus MC Fellowship 2017, and Erasmus MC Human Disease Model Award 2018. This work was supported by JSPS and MEXT KAKENHI grants (JP16H04753 and JP17H06422), and grants from AMED (20ek0109418h0002) and MHLW of Japan. Some families were collected as part of the SYNaPS Study Group collaboration funded by the Welcome Trust and strategic award (Synaptopathies) funding (WT093205 MA and WT104033AIA). This research was conducted as part of the Queen Square Genomics group at University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. Family 13 was identified in the CAUSES Study, funded by British Columbia Children’s Hospital Foundation and Genome BC.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.M.C., Y. Murakami, T.K. Formal analysis: C.T-L., Y. Murakami, T.T.M.N., Y. Maki. Funding acquisition: P.M.C., T.S.B., Y. Murakami, T.K., H.H., A.L. Investigation: R. Maroofian, T.T.M.N., E.G.K., S.K., F. Akbar, S.I., B.A., M.D., F. Ashrafzadeh, M.B., S. Efthymiou, M.C., T.S., R.L.L., H.M.M., R.T., S.M., J.S.C., K.B., F.Y.I., M.S.P., A.L, A.C.E., A.N., M.A.W., S.M.A, Y. Maki, R. Sachdev, R. Macintosh, E.E.P., G.M.S.M., T.S.B., R. Steinfeld, C.T.R., G.M.S., M.W., S.B.W., U.K., A.F.B., K.L.S., N.I., S.E.I., D.B., K.N., S.D.M., S.E.A. Methodology: P.M.C., Y. Murakami. Project administration: C.T-L., P.M.C., Y. Murakami. Resources: P.M.C., Y. Murakami, T.K., R. Maroofian, E.G.K., S.K., F. Akbar, S.I., B.A., M.D., F. Ashrafzadeh, M.B., T.S., R.L.L., H.M.M., R.T., S.M., J.S.C., K.B., F.Y.I., M.S.P., A.L, A.C.E., M.A.W., S.M.A, R. Sachdev, R. Macintosh, E.E.P., G.M.S.M., T.S.B., R. Steinfeld, C.T.R., G.M.S., M.W., S.B.W., U.K., A.F.B., K.L.S., N.I., S.E.I., D.B., K.N., S.D.M., S.E.A., H.H. Supervision: P.M.C., Y. Murakami. Visualization: C.T-L., Y. Murakami. Writing—original draft: C.T.-L. Writing—review & editing: C.T-L., P.M.C., Y. Murakami, T.K., R. Maroofian, M.W., E.E.P., T.S.B., R. Steinfeld, G.M.S.M.
Corresponding authors
Ethics declarations
Ethics Declaration
Informed written consent was obtained for every participant either from themselves or their parents. Ethical approval was granted by Centre Hospitalier Universitaire Sainte-Justine Research Center and Sick Kids Hospital. Consent was received for the use of identifying patient photographs.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Tremblay-Laganière, C., Maroofian, R., Nguyen, T.T.M. et al. PIGG variant pathogenicity assessment reveals characteristic features within 19 families. Genet Med 23, 1873–1881 (2021). https://doi.org/10.1038/s41436-021-01215-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01215-9
