
Abstract
Purpose
Germline pathogenic variants in the exonuclease domain (ED) of polymerases POLE and POLD1 predispose to adenomatous polyps, colorectal cancer (CRC), endometrial tumors, and other malignancies, and exhibit increased mutation rate and highly specific associated mutational signatures. The tumor spectrum and prevalence of POLE and POLD1 variants in hereditary cancer are evaluated in this study.
Methods
POLE and POLD1 were sequenced in 2813 unrelated probands referred for genetic counseling (2309 hereditary cancer patients subjected to a multigene panel, and 504 patients selected based on phenotypic characteristics). Cosegregation and case–control studies, yeast-based functional assays, and tumor mutational analyses were performed for variant interpretation.
Results
Twelve ED missense variants, 6 loss-of-function, and 23 outside-ED predicted-deleterious missense variants, all with population allele frequencies <1%, were identified. One ED variant (POLE p.Met294Arg) was classified as likely pathogenic, four as likely benign, and seven as variants of unknown significance. The most commonly associated tumor types were colorectal, endometrial and ovarian cancers. Loss-of-function and outside-ED variants are likely not pathogenic for this syndrome.
Conclusions
Polymerase proofreading–associated syndrome constitutes 0.1–0.4% of familial cancer cases, reaching 0.3–0.7% when only CRC and polyposis are considered. ED variant interpretation is challenging and should include multiple pieces of evidence.
Similar content being viewed by others

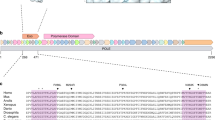
INTRODUCTION
Germline missense pathogenic variants in the exonuclease domain (ED) of polymerases epsilon (POLE; a.a. 268–471) and delta (POLD1; a.a. 304–533), which affect the proofreading capabilities of these polymerases, predispose to multiple colorectal adenomas and carcinomas, causing the so-called polymerase proofreading–associated polyposis (PPAP) (MIM 615083; 612591).1,2,3,4,5,6,7 Evidence of extracolonic tumors has been reported, including endometrial, brain, breast, ovarian, stomach, pancreas, and skin tumors, among others.2,6,7,8,9,10
Somatic pathogenic variants in the ED of POLE have been identified in 2–8% of colorectal cancer (CRC),11,12,13,14 7–15% of endometrial tumors,15,16,17,18 and more rarely in other tumor types.19,20,21 Somatic pathogenic variants affecting POLD1 ED are extremely rare. Tumors with somatic POLE ED pathogenic variants and tumors developed in the context of PPAP exhibit a dramatically increased mutation rate known as ultramutated tumor phenotype,17,21,22,23 characterized by a specific mutation signature typified by C:G→A:T transversions (TCT context) and C:G→T:A transitions (TCG context).12,24 This corresponds to signature 10 in the COSMIC Signatures of Mutational Processes in Human Cancer, or to signatures 14 and 20 if the POLE or POLD1 pathogenic variant coexists with mismatch repair (MMR) deficiency, respectively.12,24,25 Cancer patients with somatic POLE ED pathogenic variants show excellent prognosis and good response to immune checkpoint inhibition, probably due to the immune response elicited by these tumors as a result of the large number of neopeptides generated as consequence of hypermutation.26
To assess the prevalence of POLE and POLD1 pathogenic variants in hereditary cancer and refine the tumor spectrum of the associated clinical syndrome, we studied a prospective cohort of 2309 unrelated hereditary cancer patients subjected to a multigene hereditary cancer panel, and a retrospective cohort of 504 unrelated cancer patients—hereditary CRC and polyposis patients excluded—selected based on previous reports of extracolonic manifestations in PPAP, which include breast and ovarian cancer, endometrial, brain, or skin cancer, among other tumors, alone or in combination with other tumor types or colonic polyps. Cosegregation analyses, yeast-based assays, and tumor mutational analyses were performed to facilitate variant interpretation.
MATERIALS AND METHODS
Ethics statement
Patients signed consent forms and were assessed at genetic counseling units at Catalan Institute of Oncology, Vall d’Hebron Institute of Oncology, and Santa Creu i Sant Pau, Parc Taulí and Manresa Hospitals (Catalonia, Spain). The study received the approval of IDIBELL Ethics Committee.
Patients
Retrospective cohort
Five hundred and four unrelated cancer patients were analyzed, including 192 high risk breast and/or ovarian families, 178 patients with personal or familial history of different tumor types previously associated with PPAP (combinations include CRC and associated tumors, breast/ovarian cancer, skin cancer or brain), 30 patients with aggregation of other multiple tumors, either in the patient or the family, and finally, 104 patients fulfilling the criteria for TP53 genetic testing. None of the patients carried germline pathogenic variants in the known high-penetrance cancer genes associated with the patient and/or family’s phenotype (Supplementary Table S1).
Prospective cohort
Two thousand three hundred and nine unrelated familial/early-onset cancer patients, prospectively recruited from 2015 to 2018 in a hereditary cancer clinic–based context, were subjected to a multigene hereditary cancer panel.27,28 The cohort comprised patients with personal and/or familial history of breast cancer (n = 884), ovarian cancer (n = 317), and breast and ovarian cancer (n = 267); 247 familial (attenuated and classic) adenomatous polyposis cases; 354 Amsterdam- or Bethesda-positive hereditary nonpolyposis CRC cases; 15 Li–Fraumeni–suspected cases; and 225 patients with suspicion of other minor cancer syndromes.
Details of the cohorts are included in Fig. 1 and Supplementary Table S1.

BC breast cancer, BrC brain tumor, CRC colorectal cancer, EC endometrial cancer, GC gastric cancer, HBC hereditary breast cancer, HBOC hereditary breast and ovarian cancer, HOC hereditary ovarian cancer, HNPCC hereditary nonpolyposis colorectal cancer, LF Li–Fraumeni syndrome, LP likely pathogenic, OC ovarian cancer, PPAP polymerase proofreading–associated polyposis, VUS variant of unknown significance.
Mutational screening
In the retrospective cohort, direct automated (Sanger) sequencing was used to sequence exons 8–13 (+/−20 b) of POLD1 and exons 9–14 (+/−20 b) of POLE, which contain the sequences coding for the ED of each polymerase. Primer sequences were previously described.7 The hereditary cancer multigene panel applied to the prospective cohort includes the complete coding sequence of POLE and POLD1.27,28 Nonsynonymous variants located within coding exons or variants affecting canonical splice sites with a population minor allele frequency (MAF) <1% were considered.
In silico predictions
The pathogenicity of the identified missense variants was analyzed by using the metapredictor REVEL, which combines pathogenicity predictions and conservation information obtained from 18 individual scores, and provides optimal specificity and sensitivity results.29,30 REVEL score ≥0.35 was used for pathogenicity in the current study, a lower cutoff than the one recommended for clinical purposes (≥0.5) and defined based on the scores of known ED pathogenic variants (Supplementary Table S2).
Functional assessments for variant interpretation and classification
The exonuclease repair ability of POLE in the presence of ED missense variants was tested in a Schizosaccharomyces pombe system. Exome sequencing was performed on DNA extracted from the tumors of carriers of POLE/D1 variants to assess the presence of hyper or ultramutation (10 or 100 Mut/Mb respectively) and/or the mutational signature(s) associated with the presence of an ED pathogenic variant.24,31 POLE and POLD1 variant frequencies in CRC and controls were obtained from a Spanish population-based case–control study (MCC-Spain, www.mccspain.org). ED variants were classified by applying the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines32 (Supplementary Table S3). Additional details are in Supplementary Methods.
RESULTS
Of the 2309 unrelated probands included in the prospective cohort, 374 (16.2%) carried (likely) pathogenic germline variants in known cancer-predisposing genes (Fig. 1; Supplementary Table S4).
A total of 12 novel or rare (MAFgnomAD<1%) missense variants located within the EDs of POLE (n = 7) or POLD1 (n = 5) were identified in 15 families (Table 1). Five loss-of-function (LoF) variants were found in five families, and 23 predicted-deleterious POLE (n = 19) and POLD1 (n = 4) variants outside the ED were identified in 29 probands (Supplementary Fig. S1).
Mutational screening of the region coding the ED of POLE and POLD1 in the retrospective cohort (n = 504 unrelated probands) identified a missense POLE ED variant, c.1138G>T (p.Gly380Cys), and a frameshift variant, POLE c.1185_1188del (p.Glu396Thrfs*15).
Phenotypic characteristics of carriers of ED variants
Twelve rare or novel missense variants located within the EDs of POLE (n = 7) and POLD1 (n = 5) were identified in 16 unrelated individuals of the 2813 cancer patients studied (Table 1; pedigrees in Supplementary Figs. S2 and S3).
POLE c.861T>A (p.Asp287Glu) (MAFgnomAD_NFE = 0.17%) was identified in two unrelated breast cancer patients, one of whom was also diagnosed with an MMR-deficient CRC (Table 1). This variant had been previously reported in three unrelated families diagnosed with melanoma at ages 22–73 and other tumors, including breast cancer, squamous cell carcinoma, and non-Hodgkin lymphoma,10,33 and in a patient with a Lynch syndrome–associated MLH1-deficient colorectal tumor (Supplementary Table S5).34 Of the 13 studied cancer-affected individuals (in this and previous reports) only 7 carried POLE c.861T>A. No association with cancer was detected for this variant in a Spanish case–control study, either for CRC or breast cancer (source: MCC-Spain; Supplementary Table S6).
The novel, predicted pathogenic POLE c.881T>G (p.Met294Arg) variant, which affects a highly conserved amino acid, was identified in two families. One proband was a woman diagnosed with ovarian and endometrial tumors at age 36. Her mother, diagnosed with CRC and endometrial cancer at ages 42 and 55 respectively, and her CRC-affected uncle, were also carriers. POLE c.881T>G was also identified in a patient diagnosed with an MMR-proficient CRC at age 37, and in his maternal uncle, affected with three metachronous CRCs. The variant has been recently reported in a patient affected with multiple colonic adenomas, breast cancer, and endometrial cancer, diagnosed at 48, 50, and 55 years old respectively, and in her sister, with seven colonic adenomas at age 58.35
POLE c.919A>G (p.Ile307Val), affecting a conserved residue and not reported in public databases, was identified in an individual with four melanomas diagnosed at age 81 and in two of his sisters, diagnosed with melanoma at 55 and breast cancer at 83.
POLE c.1007A>G (p.Asn336Ser) (MAFgnomAD_NFE = 0.02%) was identified in two families. One of the probands, of Moroccan origin, was diagnosed with rectal cancer at age 38, and the other one with melanoma at 48. This variant is relatively frequent in African populations (MAF = 2.6%), where a total of 10 homozygous carriers have been identified among 11,975 Africans (gnomAD v.2.1.1).
POLE c.1138G>T (p.Gly380Cys) (MAFgnomAD_NFE = 0.005%), predicted deleterious, was identified in a woman diagnosed with two synchronic breast tumors, and in an unrelated individual with multiple (10–20) adenomatous polyps and no familial cancer history.
POLE c.1274A>G (p.Lys425Arg) (MAFgnomAD_NFE = 0.0025%) was identified in a woman affected with breast, ovarian, and endometrial tumors before age 50. This predicted pathogenic variant affects a highly conserved residue located within the Exo IV motif active site. The variant had been previously reported in six families, including a melanoma patient,10 a patient with early-onset CRC,3 an individual with colonic polyps and family history of CRC,36 a 63-year-old CRC patient,35 a woman diagnosed with breast cancer at age 31,35 and two first-degree relatives, who also carried the MSH2 c.942+3A>T pathogenic variant, diagnosed with three synchronic CRCs at age 30 and one CRC at age 33 respectively.35
POLE c.1277C>T (p.Ala426Val) (MAFgnomAD_NFE = 0.0034%), affecting a highly conserved residue within the Exo IV motif active site, was identified in a male diagnosed with CRC and multiple adenomas at age 44 and with renal cancer at 49, and in his CRC-affected paternal uncle. The proband’s father, an obligate carrier, was diagnosed with CRC at age 60. All three relatives also carried a likely pathogenic variant in CHEK2, c.593–1G>T (rs786203229), a gene that confers a moderate risk of breast cancer, and possibly other tumor types, including CRC.37,38 In this family, the carriers of the two variants did not develop particularly aggressive phenotypes, two of them having developed CRC late in life (ages 60 and 73).
The five ED variants identified in POLD1, p.Ile307Met, p.Ile325Val, p.Arg352Cys, p.Arg521Gln, and p.Arg525Trp, were predicted to be neutral (Table 1; pedigrees in Supplementary Fig. S3). POLD1 c.1562G>A (p.Arg521Gln) had been previously identified by our group in a 48-year-old CRC patient.7 POLD1 c.1573C>T (p.Arg525Trp) co-occurred with ATM c.7220C>A (p.Ser2407*; rs1555122149) in a woman affected with multiple tumors. ATM pathogenic variants confer a moderate risk of breast cancer (two to fivefold), but are not associated with bilateral breast tumors.37,38 Available studies have been unable to quantify the postulated increased risk to CRC.39 The carrier of POLD1 p.Arg525Arg and ATM p.Ser2407* developed a very aggressive phenotype, including three metachronous early-onset breast tumors (ages 35–53) and a CRC at age 69, supporting a cumulative effect of the two variants.
Functional assessment of ED variants: exonuclease repair yeast-based assay and tumor analysis
The exonuclease repair ability of POLE in the presence of missense ED variants was tested in a yeast system. The number of revertant colonies was significantly higher for p.Leu424Val (positive control), p.Asp287Glu, p.Met294Arg, p.Gly380Cys, and p.Ala426Val, compared with the wildtype (fold change increase of 7–13) (Fig. 2). No significant differences were observed for POLE p.Ile307Val, and a previous study had shown no effect for p.Asn336Ser.40 Notably, POLE p.Asp287Glu, p.Met294Arg, p.Leu424Val, and p.Ala426Val, the ones with the highest number of revertant colonies, were located within the DNA binding pocket structure (Fig. 3). All POLD1 ED variants identified in the study were predicted benign, and only two, p.Arg352Cys and p.Arg521Gln, affected conserved amino acids in yeast. Since all gathered evidence for those two was enough to classify them as likely benign (Table 1), we did not perform the yeast-based functional assay.

(a) Alignment of human POLE and their homolog in yeast (Pol2). The identified variants are highlighted in yellow (conserved residues) and the POLE p.Leu424Val, used as positive control, in green. ED is depicted in red (human: residues 268–471, yeast: residues 98–428) and its sequence motifs44 are shaded in gray. (b) Left panel: box plots showing mutation rates of ade6-485 S. pombe (number of colonies) for pol2 wildtype (WT, negative control), ED mutation-positive control (pol2-Leu425Val; C+) and the corresponding variants. Right panel: fold rate increase relative to the median number of revertants in the WT. Data obtained from two independent experiments performed in triplicate. *p value = 0.01, **p value = 0.01–0.001, and ***p value < 0.001 indicate the differences with the WT clone, and were calculated using the Mann–Whitney nonparametric test.

(a) 3D structure of POLE. (b) 3D structure of POLD1. Single-stranded DNA from the aligned bacteriophage T4 polymerase complex (PDB ID: 1NOY) is shown in yellow. Variants in the DNA binding pocket are highlighted in red.
Exome sequencing was performed in tumors developed by POLE p.Asp287Glu, p.Met294Arg, p.Ile307Val, p.Gly380Cys, p.Ala426Val, and POLD1 p.Arg521Gln carriers, including, as positive controls, tumors developed by POLE p.Leu424Val, POLD1 p.Asp316Gly, and POLD1 p.Asp316His carriers2,7 (Fig. 4; Supplementary Table S7). All positive controls showed hyper- or ultramutation (range: 14–478 Mut/Mb). POLE/D1 ED-associated signatures were detectable in the POLD1 p.Asp316His carrier’s CRC tumor (signature 10 contribution: 20.66%), but barely identifiable in the POLD1 p.Asp316Gly carrier’s liver metastasis (signature 10: 0.75%; signature 20 [POLD1 + MMR deficiency]: 2.65%) or in the POLE p.Leu424Val carrier’s oligodendroglioma (signature 14 [POLE + MMR deficiency]: 4%), where MMR deficiency hoarded most of the mutational signatures’ contribution.

(a) Tumor features including mismatch repair (MMR) deficiency status and mutational burden (hypermutation was considered when the tumor had more than 10 exonic Mut/Mb). aHigh-grade serous ovarian cancer. bIntestinal origin. cMicrosatellite instability (MSI) classification using Bethesda panel. C+, positive controls, i.e., tumors from carriers of variants affecting the POLE/POLD1 ED previously classified as pathogenic. (b) Mutational signature contribution (DeconstructSigs) for hyper/ultramutated tumors (>10 Mut/Mb). CRC colorectal cancer.
Three primary tumors—one MMR-deficient ovarian cancer, one MMR-deficient endometrial cancer and one MMR-proficient CRC—from two carriers of POLE p.Met294Arg, were analyzed. All three displayed hyper/ultramutation (10.96–300.85 Mut/Mb). Signature 10 was identified in the CRC (contribution: 23%), and signature 14, linked to the co-occurrence of MMR deficiency and POLE ED pathogenic variant in the ovarian tumor (contribution: 15.32%). MMR-deficient associated signatures were the major contributors in the ovarian and endometrial tumors (Fig. 4; Supplementary Table S7).
While the breast tumor from the POLE p.Gly380Cys carrier showed no hypermutation (0.67 Mut/Mb), a breast tumor from a POLE p.Asp287Glu carrier, a melanoma from a POLE p.Ile307Val carrier, the MMR-proficient CRC from the POLE p.Ala426Val and CHEK2 c.593–1G>T carrier, and a CRC from a POLD1 p.Arg521Gln carrier showed hypermutation but none of the POLE/D1-associated mutational signatures (Fig. 4; Supplementary Table S7).
To help characterize the identified variants, we used the TCGA and COSMIC tumor sequencing data to see if those variants, when occurring somatically and in absence of additional somatic POLE/D1 ED variants, caused a hyper/ultramutated phenotype and the POLE/D1 ED-associated signatures. Considering the ED variants identified in the study, only two, POLD1 p.Arg352Cys and p.Arg525Trp, fulfilled the abovementioned conditions. A CRC and a lymphoid neoplasm harboring POLD1 p.Arg352Cys, as well as a stomach carcinoma with POLD1 p.Arg525Trp, displayed hypermutation but not the POLE/D1 ED-associated signatures (Supplementary Table S8). In the case of the CRC and the stomach carcinoma, the hypermutation detected was due to the presence of microsatellite instability (MSI) (MMR deficiency signatures’ contribution: 77%), and to the presence of BRCA2 p.Pro1088Leufs*16 (BRCA1/2 signature contribution [3]: 52%), respectively. In the POLD1 p.Arg352Cys hematologic tumor, in absence of DNA repair alterations, no POLE/D1 ED-associated signatures were identified.
ED variant classification
Sixteen (16/2813; 0.57%) probands carried novel or rare ED missense variants. Taking into account several lines of evidence (functional data, tumor mutation burden and signatures, in silico predictors, cosegregation, and case–control data), and applying the ACMG/AMP guidelines for variant classification, POLE p.Met294Arg was classified as pathogenic; POLE p.Asp287Glu, POLE p.Asn336Ser, POLD1 p.Arg352Cys, and POLD1 p.Arg521Gln as likely benign; and POLE p.Ile307Val, p.Gly380Cys, p.Lys425Arg, and p.Ala426Val, and POLD1 p.Ile307Met, p.Ile325Val, and p.Arg525Trp as variants of unknown significance (VUS) (Table 1). Details of the criteria considered for the classification of each variant are shown in Supplementary Table S3.
Germline loss-of-function POLE and POLD1 variants
Six LoF heterozygous variants were identified in the retrospective (n = 1) and prospective (n = 6) cohorts: POLE c.1185_1188delGGAG (p.Glu396Thrfs*15), c.4480C>T (p.Gln1494*), and c.2297_2298insA (p.Tyr766*); and POLD1 c.230delC (p.Pro77Leufs*92), c.1195C>T (p.Gln399*), and c.3305delC (p.Pro1102Leufs*22) (Supplementary Table S9). The most prevalent phenotype in the carrier families was breast cancer (also the most represented tumor type in the studied cohorts), diagnosed in eight carriers (five families) (pedigrees: Supplementary Fig. S4). The POLD1 p.Gln399* carrier also carried a BRCA1 pathogenic variant (exon 8–13 deletion).
POLE c.1185_1188delGGAG (p.Glu396Thrfs*15), novel and disrupting the ED, perfectly segregated with breast cancer in the carrier family: the variant was present in the proband, her mother, and two maternal aunts, all four of whom were diagnosed with breast cancer (ages at diagnosis: 28, 71, 64, and 67, respectively). Two primary breast tumors from two carriers of POLE p.Glu396Thrfs*15 were analyzed by exome sequencing. None of the tumors showed hypermutation (1.09–1.70 Mut/Mb) or a POLE-associated signature (Fig. 4; Supplementary Table S7). Similar results were observed when analyzing two CRC tumors with a somatic variant in that same residue (Supplementary Table S8). No additional POLE or POLD1 somatic pathogenic variants were detected in the tumors, discarding a somatic second hit.
Tumor exome sequencing performed in the MMR-deficient colon cancer developed by the POLD1 c.230delC (p.Pro77Leufs*92) carrier revealed that ~85% of the signatures’ contribution corresponded to MMR deficiency and no representation of POLE/D1-associated signatures (Fig. 4; Supplementary Table S7). Sequencing data from a breast tumor harboring a somatic frameshift variant in the same residue as the germline POLD1 c.3305delC (p.Pro1102Leufs*22) showed neither hypermutation nor the POLE/D1-associated mutational signature (Supplementary Table S8).
The frequency of LoF variants in familial cancer (and all the subtypes considered separately) is almost identical to that observed in cancer-free controls (0.22% [5/2309] in cases vs. 0.20% [119/59,095] in controls, p value = 0.81), thus agreeing with their lack of association with cancer predisposition41 (LoF variants present in the European population data set [gnomAD] are detailed in Supplementary Table S10). Moreover, tumor sequencing demonstrated no increased mutation burden, supporting intact DNA repair.
Variants identified outside the ED
We identified 142 missense variants outside the ED (POLE n = 92 and POLD1 n = 50) in 354 patients, with no relative overrepresentation among any of the phenotype-based groups. Four POLD1 and 19 POLE outside-ED variants, identified in 29 individuals, were predicted deleterious (Supplementary Table S11).
Cosegregation data suggested that POLE c.671A>G (p.Tyr224Cys), the closest to the ED (44 amino acids upstream), was not associated with the increased breast cancer risk observed in the carrier family (Supplementary Table S11); observation supported by a yeast functional assay that revealed no mutator effect (Fig. 2). No cosegregation analyses could be performed for the remaining outside-ED predicted pathogenic variants. Tumors harboring the same somatic outside-ED missense variants in absence of other (likely) pathogenic POLE variants did not have ED-associated mutational signatures. Eight of the 23 predicted pathogenic variants (41/142 of all variants) could be analyzed in different tumor types with this approach (Supplementary Table S8). The frequency of outside-ED predicted pathogenic variants in familial cancer (and all the subtypes considered separately) does not differ from that observed in cancer-free controls (1.26% [29/2309] in cases vs. 0.91% [539/59,095] in controls, p value = 0.10) (Supplementary Table S12).
Tumor spectrum and prevalence of germline POLE and POLD1 ED variants in hereditary cancer
Two hereditary cancer families (2/2813; 0.07%) carried a pathogenic variant in POLE (p.Met294Arg), making a total of 5 carriers. The phenotypic spectrum of the carriers included (1) CRC in absence of adenomatous polyposis, diagnosed in 4 of the 5 carriers (80%) (mean age at CRC diagnosis 51; range 37–57), one of whom had developed three metachronous colorectal tumors; (2) endometrial cancer, diagnosed in two carriers (40%) (mean age at diagnosis 46; range 36–55); and (3) ovarian cancer, diagnosed in one carrier (20%) (age at diagnosis 36). Three ED variant carriers (60%) developed ≥2 primary tumors. The prevalence of (likely) pathogenic ED variants, being of 0.09% (2/2309) in the prospective familial cancer cohort, reached 0.33% (2/601) when restricted to CRC and polyposis phenotypes (Fig. 1; Supplementary Table S13).
The frequency of rare ED missense variants—including pathogenic and VUS—in POLE and POLD1 in hereditary cancer was 0.39% (9/2309). Almost half of the pathogenic variants and VUS (4/9; 44.44%) were found in patients with a referral of hereditary nonpolyposis CRC or polyposis, who accounted for 26% (601/2309) of the total number of patients tested. In this group of patients, the prevalence of ED variants increased to 0.67%. With regard to the most represented group of patients, i.e., those referred for genetic testing as hereditary breast and/or ovarian cancer, the frequency of ED variants was 0.27% (4/1468) (Fig. 1; Supplementary Table S13).
DISCUSSION
POLE and POLD1 ED variant interpretation is challenging and the inclusion of multiple pieces of evidence is highly advisable. Population and cosegregation data, in silico prediction of pathogenicity, location within the DNA binding cleft, results of the functional repair assay, and tumor mutational data were taken into consideration for variant classification. Based the relevance of each piece of evidence, we adapted the ACMG/AMP variant classification guidelines to the particular conditions of POLE and POLD1 ED variants and to the relevance of the functional information gathered, i.e., tumor mutational signatures, yeast repair assay results, and alteration of the DNA binding pocket (Table S3).
We consider tumor sequence analysis of utmost importance for a proper classification of POLE and POLD1 ED variants. Mutational signature 10, in the presence of hyper/ultramutation, is unequivocally linked to the presence of an ED pathogenic variant—a priori independently of the tumor type analyzed—however, one must be certain of the inexistence of a somatic ED pathogenic variant that could be causing the presence of the POLE/D1-associated tumor molecular phenotype. Moreover, a challenging situation occurs when the tumor we analyze has other DNA repair defects, e.g., MMR deficiency (MSI). In this situation, if the mutational signature analysis does not reveal any of the POLE/D1-associated signatures, i.e., signatures 14 or 20 when the germline variant affects the ED of POLE or ED respectively, it is highly advisable to analyze a second tumor, ideally MMR proficient, developed in the same family or harboring the same variant but in a somatic context, verifying that no other somatic ED variant is present.
Due to the high homology of the ED of POLE and POLD1 in human and yeast, this model has been widely used for variant functional assessment. In our study, unequivocal repair impairment (p < 0.001) was observed for POLE p.Leu424Val (positive control), p.Met294Arg (likely pathogenic), and p.Ala426Val (uncertain significance), but also for p.Asp287Glu, a variant classified as likely benign based on cosegregation data, frequency in cases and controls, and absence of an ED-associated mutational signature. It seems evident that, in addition to the (likely) pathogenic variants directly affecting the exonuclease catalytic residues, such as POLD1 p.Asp316Gly, p.Asp316His, ED variants affecting residues in direct contact or in very close proximity to the DNA fragment, i.e., in the DNA binding pocket, have a very high chance of being pathogenic. In fact, the vast majority of POLE and POLD1 (likely) pathogenic variants reported to date are localized in the DNA binding cleft, including POLE p.Thr278Lys, p.Met294Arg (this study), p.P286L, p.N363K, p. D368N, p.Leu424Val, and p.Pro436Ser, as well as POLD1 p.Leu474Pro and p.Ser478Asn (Supplementary Table S2). Hamzaoui et al. observed that variants interfering with DNA binding (p.P286L and p.N363K) produce a higher mutagenic effect in yeast than variants disrupting ion metal coordination at the exonuclease site.35 In line with Barbari et al.,15,40 our results indicate that any variant located in the DNA binding cleft shows a mutator effect in yeast, even if it is not pathogenic, as occurs with p.Asp287Glu. Based on this and the high variability of the yeast assay among replicates and experiments, also observed in other studies,35,40 we have used its results with caution. We posit that yeast assay observations may be considered supporting or, at most, moderate (not strong) evidence for ACMG/AMP variant classification, depending on the level of significance when compared with the wildtype clone.
In our study, two families carrying POLE and POLD1 VUS also carried pathogenic variants in ATM and CHEK2, two genes that confer a moderate risk of breast cancer.37,38 The aggressive phenotype developed by the POLD1 p.Arg525Arg and ATM (p.Ser2407*) carrier supports a cumulative effect of the two variants, while that may not be the case for the carriers of POLE p.Ala426Val (VUS) and CHEK2 c.593–1G>T, where two of the three carriers developed late-onset CRC. Hamzaoui et al. identified two relatives who carried both POLE p.K425R (VUS) and MSH2 c.942+3A>T (pathogenic), in a family with multiple CRC-affected members diagnosed at extremely young ages (range: 19–33), suggesting a cumulative effect of the two variants.35 Whether the aggregation of cancer in these families is explained only by the pathogenic variants in ATM, CHEK2, or MSH2, or the combined effect with the POLE/D1 variants (all of them classified as VUS until now), remains to be elucidated.
The role of LoF and outside-ED POLE and POLD1 heterozygous variants has been a matter of controversy since the description of PPAP in 2013. Our data suggest that these variants are most likely nonpathogenic, at least not associated with PPAP—this being relevant to avoid misdiagnoses in the clinical setting. A particular case is that of POLE p.Glu396Thrfs*15, identified in four breast cancer–affected women of the same family in absence of additional pathogenic variants in known or candidate hereditary cancer genes (exome sequencing; data not shown). Despite the absence of tumor hypermutation and POLE-associated mutational signatures, whether the variant is the cause of the cancer aggregation in the family remains to be elucidated, and if so, what is the molecular mechanism underlying its potential causal effect.
In conclusion, our findings indicate that PPAP constitutes 0.1–0.4% of familial cancer cases referred to a hereditary cancer clinic, 0.3–0.7% when considering nonpolyposis CRC and polyposis patients, with the most commonly associated tumor types being colorectal, endometrial, and ovarian cancers. ED variant interpretation is challenging and should include multiple pieces of evidence. LoF and outside-ED variants are not associated with PPAP, a cancer-predisposing syndrome characterized by a defect in the proofreading activity of polymerases epsilon and delta.
References
Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144.
Valle L, Hernández-Illán E, Bellido F, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014;23:3506–3512.
Rohlin A, Eiengård F, Lundstam U, et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes Chromosomes Cancer. 2016;55:95–106.
Elsayed FA, Kets CM, Ruano D, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet. 2015;23:1080–1084.
Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137:320–331.
Chubb D, Broderick P, Frampton M, et al. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J Clin Oncol. 2015;33:426–432.
Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18:325–332.
Hansen MF, Johansen J, Bjørnevoll I, et al. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam Cancer. 2015;14:437–448.
Rohlin A, Zagoras T, Nilsson S, et al. A mutation in POLE predisposing to a multi-tumour phenotype. Int J Oncol. 2014;45:77–81.
Aoude LG, Heitzer E, Johansson P, et al. POLE mutations in families predisposed to cutaneous melanoma. Fam Cancer. 2015;14:621–628.
Brannon AR, Vakiani E, Sylvester BE, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014;15:454.
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337.
Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664.
Giannakis M, Mu XJ, Shukla SA, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;17:1206.
Barbari SR, Shcherbakova PV. Replicative DNA polymerase defects in human cancers: consequences, mechanisms, and implications for therapy. DNA Repair (Amst). 2017;56:16–25.
Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Church DN, Briggs SE, Palles C, et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22:2820–2828.
Church DN, Stelloo E, Nout RA, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst. 2015;107:402.
Shinbrot E, Henninger EE, Weinhold N, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24:1740–1750.
Zou Y, Liu FY, Liu H, et al. Frequent POLE1 p.S297F mutation in Chinese patients with ovarian endometrioid carcinoma. Mutat Res. 2014;761:49–52.
Erson-Omay EZ, Çağlayan AO, Schultz N, et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. 2015;17:1356–1364.
Campbell BB, Light N, Fabrizio D, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell. 2017;171:1042–.e1010.
Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230:148–153.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421.
Haradhvala NJ, Kim J, Maruvka YE, et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun. 2018;9:1746.
Bourdais R, Rousseau B, Pujals A, et al. Polymerase proofreading domain mutations: new opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit Rev Oncol Hematol. 2017;113:242–248.
Feliubadaló L, Tonda R, Gausachs M, et al. Benchmarking of whole exome sequencing and ad hoc designed panels for genetic testing of hereditary cancer. Sci Rep. 2017;7:37984.
Castellanos E, Gel B, Rosas I, et al. A comprehensive custom panel design for routine hereditary cancer testing: preserving control, improving diagnostics and revealing a complex variation landscape. Sci Rep. 2017;7:39348.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885.
Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017;18:225.
Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24:107–113.
Jansen AM, van Wezel T, van den Akker BE, et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet. 2016;24:1089–1092.
Hamzaoui N, Alarcon F, Leulliot N, et al. Genetic, structural, and functional characterization of POLE polymerase proofreading variants allows cancer risk prediction. Genet Med. 2020 May 19; https://doi.org/10.1038/s41436-020-0828-z [Epub ahead of print].
Shirts BH, Casadei S, Jacobson AL, et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016;18:974–981.
Couch FJ, Shimelis H, Hu C, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–1196.
Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–2257.
West AH, Blazer KR, Stoll J, et al. Clinical interpretation of pathogenic ATM and CHEK2 variants on multigene panel tests: navigating moderate risk. Fam Cancer. 2018;17:495–505.
Barbari SR, Kane DP, Moore EA, Shcherbakova PV. Functional analysis of cancer-associated DNA polymerase ε variants in Saccharomyces cerevisiae. G3 (Bethesda). 2018;8:1019–1029.
Mur P, Palles C, Tomlinson I, Valle L. Reply to: “Development of an MSI-positive colon tumor with aberrant DNA methylation in a PPAP patient”. J Hum Genet. 2020;65:513–514.
Li Q, Wang K. InterVar: clinical Interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–280.
Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2015;18:325–332.
Shevelev IV, Hübscher U. The 3’ 5’ exonucleases. Nat Rev Mol Cell Biol. 2002;3:364–376.
Acknowledgements
We thank Gemma Aiza, Mireia Menéndez, Sara González, and Xavier Muñoz for support, and Biobanc HUB-ICO-IDIBELL, integrated in the Spanish Platform Biobanks Network and funded by Instituto de Salud Carlos III (PT17/0015/0024) and Xarxa Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncologia de Catalunya. This work was funded by the Spanish Ministry of Science and Innovation, cofunded by FEDER funds (SAF2016-80888-R, SAF2015-68016-R, Severo Ochoa SVP-2014-068895 contract [L.M.-P.]); Instituto de Salud Carlos III (PI16/00563, PI16/00588, PI14/00613, PI19/00553, CIBERONC CB16/12/00234, CIBERESP, Sara Borrell contract [P.M.]); Government of Catalonia [AGAUR 2017SGR1282, CERCA Program]; and Fundación Olga Torres. This study was facilitated by COST Action CA17118, supported by COST (European Cooperation in Science and Technology).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Senior co-authorship: Conxi Lázaro, Laura Valle
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mur, P., García-Mulero, S., del Valle, J. et al. Role of POLE and POLD1 in familial cancer. Genet Med 22, 2089–2100 (2020). https://doi.org/10.1038/s41436-020-0922-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0922-2
