
Abstract
Purpose
Congenital diaphragmatic hernia (CDH) is associated with significant mortality and long-term morbidity in some but not all individuals. We hypothesize monogenic factors that cause CDH are likely to have pleiotropic effects and be associated with worse clinical outcomes.
Methods
We enrolled and prospectively followed 647 newborns with CDH and performed genomic sequencing on 462 trios to identify de novo variants. We grouped cases into those with and without likely damaging (LD) variants and systematically assessed CDH clinical outcomes between the genetic groups.
Results
Complex cases with additional congenital anomalies had higher mortality than isolated cases (P = 8 × 10−6). Isolated cases with LD variants had similar mortality to complex cases and much higher mortality than isolated cases without LD (P = 3 × 10−3). The trend was similar with pulmonary hypertension at 1 month. Cases with LD variants had an estimated 12–17 points lower scores on neurodevelopmental assessments at 2 years compared with cases without LD variants, and this difference is similar in isolated and complex cases.
Conclusion
We found that the LD genetic variants are associated with higher mortality, worse pulmonary hypertension, and worse neurodevelopment outcomes compared with non-LD variants. Our results have important implications for prognosis, potential intervention and long-term follow up for children with CDH.
Similar content being viewed by others

INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a developmental defect of the diaphragm present at birth, occurring in ~3 per 10,000 live births. Improvements in early diagnosis and clinical treatment of CDH have led to improved survival; however, CDH is still associated with significant mortality, long-term morbidity, and developmental disabilities in some individuals. Approximately 15–20% of individuals with CDH have an identifiable genetic cause,1,2,3,4 including de novo single-nucleotide variants (SNVs) and small insertions or deletions (indels) that affect a single gene,5,6 as well as de novo copy-number variants (CNVs) that delete or duplicate contiguous genes.7,8 Pulmonary hypertension (PH) resulting in significant morbidity and mortality and/or developmental delay are more common in CDH patients.9,10 To date, no studies have comprehensively examined the association of genetic variants with clinical outcomes in CDH patients.
Our previous studies in congenital heart disease (CHD), a common comorbid condition with CDH, have shown increased burden of de novo variants in patients with neurodevelopmental disorders.11,12 We hypothesize that damaging genetic variants that cause congenital anomalies in general often have pleiotropic effect in development and homeostasis of other organs, leading to worse clinical outcomes.
To assess whether de novo predicted deleterious variants are associated with differential clinical outcomes in CDH patients, we analyzed 462 CDH patients with parent–offspring trio sequencing enrolled at birth and prospectively followed by the Diaphragmatic Hernia Research & Exploration; Advancing Molecular Science (DHREAMS) study. We identified and classified de novo SNVs, indels, and CNVs, and hypothesized that de novo predicted deleterious variants would be associated with higher mortality and worse clinical and developmental outcomes.
MATERIALS AND METHODS
Study subjects
The participants in this study were enrolled as neonates with a radiologically confirmed diaphragm defect requiring neonatal repair in the DHREAMS study (http://www.cdhgenetics.com) from 2005 to 2018 and were followed prospectively. DHREAMS is a multicenter study of individuals with a diaphragm anomaly.5 Columbia University Irving Medical Center (CUIMC) is the central coordinating center and data and biospecimen repository.10,13 Each enrolling clinical site is a regional tertiary care hospital with a neonatal intensive care unit that includes an extracorporeal membrane oxygenation (ECMO) program.13 There is no treatment protocol in the study, and clinical management is determined by the treating clinical team. A blood or saliva specimen for genetic analysis was collected from the CDH patient and both parents (trio).
Ethics statement
All studies were approved by the institutional review boards at each participating institution and the CUIMC Institutional Review Board (IRB), and signed informed consent was obtained.
Clinical factors and outcomes
Clinical data
The clinical data collected for this cohort have been previously described.10 Briefly, clinical data were prospectively collected from medical records and entered into a central Research Electronic Data Capture (REDCap) database.14 Parental information including race, highest level of education, and occupation were collected at the time of study enrollment by a single research coordinator. The Hollingshead’s Four Factor Index of social status (HI), a measure of socioeconomic status (SES), was calculated for each household.15 Participants with only a diaphragm defect were classified as isolated CDH while participants with at least one additional congenital anomaly (e.g., congenital heart defect, central nervous system anomaly, gastrointestinal anomaly, skeletal anomaly, cleft lip/palate, etc.) were classified as complex CDH. Pulmonary hypoplasia, cardiac displacement, and intestinal herniation were considered to be part of the diaphragm defect sequence and were not considered independent malformations.
Outcomes
Data on the child’s current and past health were gathered by parental interview at two years of age by the CUIMC research coordinator. Follow up data were collected through January 2019. Echocardiograms to assess pulmonary hypertension were centrally analyzed independently by two cardiologists using a standardized protocol10 (see Supplementary Notes). For this study, postoperative PH at one month or three months was quantified as none, mild, or severe. Mortality or survival status prior to initial discharge was collected through medical record review. Growth measurements at birth and two years of age, including height and head circumference, were collected and converted to z-scores. Standardized developmental assessments, including the Bayley Scales of Infant Development, third edition (BSID-III)16 and the Vineland Adaptive Behavior Scales, second edition (VABS-II),17 were administered by child psychologists who were assessed for reliability.10 The BSID-III and VABS-II were completed either in person at a participating center, locally by certified specialists, or by telephone (VABS-II only). Both assessments were completed with interpreters if the family’s native language was not English. The VABS-II and BSID-III main domains in a standardized population have a mean score of 100 and a standard deviation (SD) of 15.
Identification of de novo variants
At the time the data was frozen for this analysis, 462 of all 647 neonatally enrolled probands met criteria and had trio sequencing data (see Supplementary Notes). The calling and annotation of de novo SNVs and indels were processed using a pipeline based on GATK Best Practice v3 followed with heuristic hard filters as described in our previous study.6 All coding SNVs and indels were classified as synonymous, missense, inframe, or likely gene disrupting (LGD, which includes frameshift indels, canonical splice site, or nonsense variants). The missense variants were further predicted as deleterious missense variants by an integrated “missense badness, PolyPhen-2, and constraint” (MPC) score ≥2 (unpublished data) and phred-scaled CADD18 v1.3 score ≥20, and plausible deleterious missense variants by MPC score between 1 and 2 and CADD score ≥20.
De novo CNVs were identified using an inhouse pipeline of read depth–based algorithm based on CNVnator19 v0.3.3 in genome sequencing trios (Figure S1, Supplementary Notes). If a sample had larger number of de novo CNV segments, i.e., more than one SD away from sample mean, the CNVs were called by the additional pair-end/split-read (PE/SR) evidence Lumpy20 v0.2.13 and genotyped by SVtyper21 v0.1.4. Only the CNVs supported by both read depth and PE/SR would be kept for downstream analysis. De novo CNVs were called with at least two confirmed evidences: (1) normalized read depth, (2) discordant pairs, (3) split reads, (4) the allelic copy ratio in terms of B allele frequency. Twenty-two of 25 (88%) reported de novo CNVs in CHD patients were confirmed by quantitative polymerase chain reaction (qPCR). We mapped de novo CNVs on GENCODE (v29) protein coding genes with at least 1 bp in the shared interval. The GENCODE genes were annotated with variant intolerance metric by gnomAD probability of loss-of-function intolerance (pLI),22 haploinsufficiency, or triplosensitivity metrics from ClinGen genome dosage map.23
Genetic groups of CDH patients
Based on the enrichment of deleterious de novo variants in CDH cases compared with controls or background mutation rate in previous studies,6,8 we defined likely damaging (LD) variants as (1) de novo LGD or deleterious missense variants in genes that are constrained (gnomAD pLI ≥0.9) and highly expressed in developing diaphragm,8 or (2) de novo LGD or deleterious missense variants in known risk genes of CDH or commonly comorbid disorders (CHD), or (3) plausible deleterious missense variants in known risk genes of CDH or commonly comorbid disorders (CHD), or (4) deletions in constrained (gnomAD pLI ≥0.9) or haploinsufficient genes from ClinGen genome dosage map, or (5) CNVs implicated in known syndromes. We classified CDH patients into two genetic groups: (1) LD, if the patient carried at least one LD variant; (2) non-LD, if the patient carried no such variants.
Statistical analysis of associations of genetic factors with clinical outcomes
We calculated all pairwise Spearman correlations of all clinical data and outcomes of CDH patients. We considered these nine clinical outcomes in the association analysis: mortality prior to initial discharge, pulmonary hypertension at 1 month and 3 months, growth (height and head circumference) at 2 years, BSID-III (language, cognitive, and motor scores) at 2 years, and VABS-II adaptive behavior composite score at 2 years.
Association tests
To estimate the difference of outcomes between LD group and non-LD group, we carried out Fisher’s exact test or Student’s t-test for binary or quantitative outcomes as appropriate. Additionally, we used Lasso-selected features as covariates to account for other clinical factors that were correlated with outcomes (see Supplementary Notes). The association tests were analyzed separately for all CDH, isolated and complex CDH cases, but with same covariates for each outcome.
Independent tests definition
Since our primary outcomes were highly correlated (Figure S5), the burden of multiple-test adjustment was smaller than the number of tests. We took an approach similar to Werling’s method24,25 to estimate the number of independent tests for multiple-test adjustment. Specifically, we performed eigenvalue decomposition on correlation matrix of all the outcomes used for association with genetic factors and counted the number of principle components (N) that combined explain 99% of the variance of the original data. We then set the Bonferroni correction threshold at 0.05/N.
Software
Statistical and graphical analyses were performed using R version 3.4.1, with R packages “glmnet,” “pcaMethods,” “sjstats,” “dplyr,” “corrplot,” “ggplot2,” “gridExtra.”
RESULTS
Sample characteristics
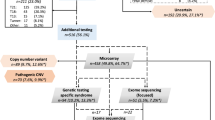
A total of 647 of approximately 840 eligible neonates born at participating centers during the study time period consented to enrollment in the DHREAMS study (Fig. 1). Of 462 patients who had genetic data, 428 cases had exome and/or genome sequencing, including 125 cases with exome sequencing, 394 cases with genome sequencing and 91 cases with both exome and genome sequencing. Thirty cases had conditions diagnosed first through clinical testing (6 of 30 also had exome sequencing), 4 cases through cytogenetic and microarray (CMA),5 and 7 cases through candidate gene test (1 of 7 also had exome sequencing) (Table S1). The other 185 patients did not have DNA on both biological parents or had insufficient DNA on the proband and had the same clinical characteristics as the 647 cases and 462 cases with genetic data (Table S2). Some participants included in this series have been previously published including CNV analyses,5 52 trios with exome sequencing, and 117 trios with genome sequencing.6 The LD group was composed of 85 participants including 43 with de novo LGD or deleterious missense variants in known risk genes or constrained genes with high diaphragm expression, 9 with plausible deleterious missense variants in known risk genes (Table S3), 7 with deletions in constrained or haploinsufficient genes (Table S4), 4 with CMA large deletions,5 and 29 genetically diagnosed subjects through clinical genetic testing. The non-LD group was composed of the other 377 participants. As reported in our previous SNV and indels analyses in CDH cases6 and CNV calls analyses in this study (Figure S3), there were no technical batch effects and the sequencing depth did not affect the variants calling.

BSID-III Bayley Scales of Infant Development third edition, CDH congenital diaphragmatic hernia, PH pulmonary hypertension, VABS-II Vineland Adaptive Behavior Scales second edition.
The participants' demographic and clinical information is summarized in Table 1. Two thirds of the participants (67%) were white, non-Hispanic, and slightly more than half (57%) were male. The diaphragm lesion was left sided for 80%, and the diaphragm defect was prenatally diagnosed for 75%. On average, participants were full term (mean 38-week gestation) with an average Apgar of 5 at 1 minute and 7 at 5 minutes. A total of 229 (35%) cases had additional congenital anomalies and were defined as complex; 394 (61%) participants had only a diaphragm defect without additional anomalies and were defined as isolated and the complexity status was unknown for 24 (4%). One third (32%) were treated with extracorporeal membrane oxygenation (ECMO) and over half (52%) received inhaled nitric oxide. Of the 566 participants who survived to surgical repair, 54% required a patch to repair the diaphragm. Two thirds required oxygen therapy at 28 days of life. Pulmonary hypertension was present for 56% of the 399 participants who were assessed at 1 month and 20% of the 306 participants who were assessed at 3 months. The cohort had 22% (144/647) mortality prior to discharge; 23 participants were still hospitalized at the time of the data freeze (1 January 2019) so discharge status was unknown. Six participants died between initial discharge and 2 years of age, and three participants died between 2 and 5 years of age.
At the time of data analysis, 275 of the 385 (71%) participants who had reached 2 years of age had a two-year developmental assessment (Figure S4, BSID-III n = 222, VABS-II n = 274). The average age at assessment was 25 months with a range of 22 to 36 months (one outlier assessment completed at 14 months). On average, the BSID-III scores for all domains and VABS-II composite scores were 6–7 points below the normal mean of 100. Growth outcomes (standardized z-scores of height and head circumference) at two years of age were completed on 249 participants. On average, the height at 2 years was 0.24 below the normal mean z-scores of 0 and was similar (0.02) to normal head circumference mean at 2 years. As expected, all measures were highly intercorrelated (Figure S5). There were in total 27 tests considering the association analyzed in all cases, isolated cases, and complex cases separately. We took eigenvalue decomposition to estimate ten effective tests based on the sum of eigenvalues that explain 99% of variation (Figure S6). The significant threshold was 5 × 10−3 correcting for multiple tests.
Higher mortality rate in complex CDH cases and CDH cases with LD variants
Of 462 CDH cases analyzed by genetic status, 31% (53/170) of complex cases and 11% (32/291) of isolated cases carried LD variants (Table S5). The overall mortality in this group was 20% before initial discharge. Complex cases had a higher mortality rate than isolated cases (31% vs. 13%, P = 8 × 10−6). Compared with the non-LD group, mortality prior to initial discharge was higher in the LD group (P = 4 × 10−4, Fig. 2a). The difference in mortality between the LD and non-LD group was largely attributable to differences in the isolated cases. The isolated cases with LD variants had a mortality rate (31%) that was similar to complex cases and was much higher than isolated cases without LD (11%, P = 3 × 10−3).

(a) Mortality prior to initial discharge. (b) Pulmonary hypertension at 1 month. (c) Pulmonary hypertension at 3 months. P value is given by Fisher’s exact test after correcting multiple tests (P < 5 × 10−3). **P < 0.001; *P < 0.005. LD likely damaging.
Higher PH prevalence in CDH cases with LD variants
A total of 335 cases with genetic data were assessed for PH at 1 month. Overall 178/335 of the cases with genetic data (53%) were diagnosed with PH at 1 month. Complex cases had a trend of higher frequency of PH at 1 month than isolated cases (62% vs. 48%, P = 1 × 10−2, Fig. 2b). LD cases had significantly higher prevalence of PH at 1 month than non-LD cases (74% vs. 49%, P = 8 × 10−4). Similar to mortality, the difference was largely driven by isolated cases: isolated cases with LD had a frequency similar to complex cases and higher than isolated cases without LD (85% vs. 44%, P = 1 × 10−3). The overall prevalence of PH at 3 months was much lower (51/268 = 19%), and had a higher prevalence in cases with LD variants remained (37% vs. 16%, P = 3 × 10−3, Fig. 2c).
LD variants were associated with worse neurodevelopmental status at two years of age
LD cases had significantly lower BSID-III language (β = 17, P = 5 × 10−5), BSID-III cognition (β = 14, P = 1 × 10−4), BSID-III motor (β = 15, P = 3 × 10−4), and VABS-II adaptive behavior composite (β = 12, P = 4 × 10−5) scores than non-LD cases (Fig. 3). There was a consistent trend of the pattern in isolated and complex cases. Adaptive behavior function from the VABS-II had the most significantly different (mean score 84 vs. 96, β = 12, P = 4 × 10−5), consistent across isolated (β = 11) and complex cases (β = 10). For all neurodevelopmental domains in both the BSID-III and VABS-II, the complex cases had worse neurodevelopmental outcomes than isolated cases in the corresponding genetic groups (Fig. 3).

(a) Bayley Scales of Infant Development third edition (BSID-III) language, (b) BSID-III cognition, (c) BSID-III motor, (d) Vineland Adaptive Behavior Scales second edition (VABS-II) composite adaptive behavior. The black line in the middle of the box is the mean value for each group. The vertical size of the box is the interquartile range (IQR). The whisker is 1.5 × IQR. P value is given by Student’s t-test with bold as significant after correcting multiple tests (P < 5 × 10−3).
Our previous studies9 showed that many different clinical factors were associated with developmental delay (Fig. 4). To account for the factors that had independent associations with outcomes, we used Lasso regularized regression to select these factors and then included them as covariates in linear regression models to test the association of LD variants with developmental outcomes. By penalizing the absolute size of the regression coefficients, the Lasso regression ends up in a situation where some of the parameter estimates may be exactly zero, which is convenient when the predictors are highly correlated26,27 (Figure S5). With Lasso-selected clinical factors as covariates, we estimated the effect size of LD variants to BSID-III and VABS-II scores. We found that the effect size of LD variants was similar to the estimates from the univariate test without clinical factors as covariates for all measures except BSID-III language. Specifically, with BSID-III language, there was still a trend toward worse score in LD patients (β = 8.5) without reaching statistical significance after correcting multiple tests (P = 9 × 10−3). Notably, families with one more point of SES normalized z-score based on parental education and occupation had 4.5 points increased scores on BSID-III language domain (β = 4.3, 95% CI = 2.2–6.4, P = 1 × 10−4), and oxygen at discharge had substantial adverse impact on BSID-III language domain scores (β = 12, 95% CI = 5.5–18, P = 3 × 10−4), consistently in both isolated and complex cases. With BSID-III cognitive score, LD patients had significantly worse outcome than non-LD patients (β = 11.4, 95% CI = 5.7–17, P = 1 × 10−4). The difference was more pronounced in complex cases (β = 16.6, 95% CI = 7.5–25.8, P = 8 × 10−4), whereas the effect size was small in isolated cases (β = 3.4, 95% CI = −11.5–4.8, P = 0.4). BSID-III cognitive domain scores also had a similar trend that associated with supplemental oxygen intake (SOI; oxygen at discharge: β = 7.0, 95% CI = 1.1–12.8, P = 2 × 10−2, and ECMO: β = 5.4, 95% CI = 0.4–10.5, P = 4 × 10−2). With BSID-III motor domain scores, LD patients had significantly lower scores (β = 14.7, 95% CI = 9.1–20.3, P = 8 × 10−7), which was consistent in isolated (β = 15.5, 95% CI = 7.3–23.7, P = 3 × 10−4) and had a similar trend in complex cases (β = 10.6, 95% CI = 2.0–19.3, P = 2 × 10−2). BSID-III motor domain scores were significantly associated with SOI (oxygen at discharge: β = 12.8, 95% CI = 6.9–18.7, P = 3 × 10−5), and the effect was consistent in both isolated and complex cases (β = 15 and 11, respectively). With VABS-II composite adaptive behavior score, patients with LD variants had significantly lower scores (β = 10.7, 95% CI = 6.5–14.8, P = 1 × 10−6), consistently in both isolated and complex cases (β = 10.2 and 8.6, respectively).

(a) Bayley Scales of Infant Development third edition (BSID-III) language, (b) BSID-III cognitive, (c) BSID-III motor, (d) Vineland Adaptive Behavior Scales second edition (VABS-II) composite adaptive behavior. Effect size is the coefficient/beta, which aligns with outcome on the y-axis, with the vertical dashed line (v = 0) separating positive and negative direction of association. Significant P value after correcting multiple tests (P < 5 × 10−3) is noted. ECMO extracorporeal membrane oxygenation, LD likely damaging variants, PH pulmonary hypertension, SES socioeconomic status.
LD cases were lower (β = 0.86 for z-score, P = 1 × 10−3, Figure S7) than non-LD cases at 2 years of age. There was no significant difference with head circumference at 2 years of age (Figures S7–S9).
After the exclusion of carriers with known syndromic abnormalities and/or large CNVs (Table S1), the association of worse clinical outcomes for LD cases compared with non-LD cases were consistent with similar effect size (Figures S9–11, see Supplementary Notes). Lastly, there was no difference in clinical outcomes and factors between clinical sites (P > 0.05), which was consistent with our prior study of the DHREAMS cohort.10 There was no statistical difference observed in mortality, need for ECMO, and percentage with pulmonary hypertension at one month of age across sites.
DISCUSSION
This study assesses the association between genetic variants and outcomes at two years of age in CDH patients. We classified 462 CDH neonates into two genetic groups: (1) CDH patients with LD variants, including de novo SNVs, indels, CNVs involving known risk genes or constrained genes with high diaphragm expression; and (2) other CDH patients. We assessed the association of these genetic classes with mortality prior to initial discharge, pulmonary hypertension, neurodevelopmental outcomes, and growth measurements at two years of age for genetic groups. We found that de novo LD variants were associated with higher mortality and prevalence of PH, worse neurodevelopmental outcomes at two years of age, but no difference in height or head circumference at two years. The associations were largely observed in both complex cases and isolated cases, and independent of other clinical factors.
The underlying hypothesis of this study is that damaging variants that cause CDH often have pleiotropic effects on the development of other parts of the body, leading to overall worse clinical and neurodevelopmental outcomes. One challenge in testing this hypothesis in this early stage of understanding CDH genetics is how to identify damaging variants when established risk genes contribute to only a small fraction of cases.1,6 Previous genetic studies of CDH6,7,8,28 and a common comorbid condition CHD11,12 showed that cases are enriched with deleterious de novo variants, especially in genes that are highly expressed in the affected organ during development,8,11 or in genes that are intolerant of variants in general population.6 Collectively, about 10–30% of CDH cases are attributable to de novo variants. Based on these observations, we defined LD variants as deleterious de novo variants in established risk genes or genes that are constrained and highly expressed in developing diaphragm. By this approach, we identified LD variants in about 20% of CDH cases. As we develop better methods to identify genes relevant to CDH development, we should be able to identify additional monogenic and oligogenic de novo and inherited variants contributing to CDH and clinical outcomes.
In our study, cases with LD variants had worse clinical and neurodevelopmental outcomes than cases without LD variants. Additionally, complex cases generally had worse outcomes than isolated cases. The effect of LD variants was consistent in both isolated and complex cases with a few exceptions, indicating that LD variants affect clinical outcome independent of the presence of additional anomalies at birth.
Previous studies indicated that abnormal pulmonary vascular development and function is a significant clinical problem in CDH infants.29 In our cohort, CDH cases with LD variants had higher PH prevalence at 1 or 3 months compared with cases without LD variants. The mechanism of PH in CDH could be in part the pleiotropic effect of genes with LD variants that lead to the premature differentiation of smooth muscle cells into a contractile phenotype.30 The PH prevalence is lower at 3 months due to early mortality in those severely affected and due to continued maturation of the pulmonary vasculature and efficacy of clinical treatments.
The neurodevelopmental scores in the BSID-III and VABS-II were assessed for correlation, especially for the similar domains. Patients with LD variants had worse motor and cognitive function, likely reflecting effects of these genes on overall brain function. The VABS-II assesses adaptive behaviors including communication, daily living skills, socialization, and motor skills, and LD patients had worse adaptive behavioral skills compared with non-LD patients. BSID-III language is correlated with SES, and this was also the case in our study. SOI, i.e., oxygen at discharge, is likely a measure of overall clinical status and likely reflects disease severity, which may be inversely correlated with the child’s opportunity to develop new skills. It is important to note that even though this is a multicenter study and clinical management was left to the discretion of each center’s clinical teams, there was no difference of clinical outcomes and factors between clinical sites (P > 0.05).
Limitations
While we report the largest prospective study of the natural history, genetic etiology, and clinical outcomes in patients with CDH, there are several limitations to our study. We did not successfully recruit all eligible participants. Our experience is that the most severe cases who died shortly after birth were less likely to enroll. Not all probands had DNA available on both parents, and therefore were not analyzed in this study although we did not observe significant differences in clinical characteristics for that group. Some patients did not complete all prospective clinical assessments, in particular the neurodevelopmental assessment, and there may be some unknown bias in what families completed those assessments. Our study is limited by the relatively young age at assessment, as some children may present with learning and/or behavioral disabilities as they get older.
Since our knowledge of CDH risk genes is incomplete and the methods to interpret deleterious missense variants are imperfect, it is likely that the groups of cases with LD or non-LD variants are not precise. Future gene discovery studies with larger sample sizes will allow us to identify damaging variants with greater accuracy and reassess the association of outcomes.
In summary, our association analysis demonstrates that CDH cases with de novo LD variants had higher mortality prior to discharge, higher prevalence of pulmonary hypertension, and worse neurodevelopmental outcomes at 2 years of age. Accurate prognostic information is critical for prenatal diagnosis and counseling and neonatal management to help the families and clinicians make informed decisions about management.
References
Yu L, Hernan RR, Wynn J, Chung WK. The influence of genetics in congenital diaphragmatic hernia. Semin Perinatol. 2020;44:151169.
Kammoun M, Souche E, Brady P, et al. Genetic profile of isolated congenital diaphragmatic hernia revealed by targeted next-generation sequencing. Prenat Diagn. 2018;38:654–663.
Slavotinek AM. The genetics of common disorders—congenital diaphragmatic hernia. Eur J Med Genet. 2014;57:418–423.
Pober BR, Lin A, Russell M, et al. Infants with Bochdalek diaphragmatic hernia: sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am J Med Genet A. 2005;138A:81–88.
Yu L, Wynn J, Ma L, et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49:650–659.
Qi H, Yu L, Zhou X, et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018;14:e1007822.
Zhu Q, High FA, Zhang C, et al. Systematic analysis of copy number variation associated with congenital diaphragmatic hernia. Proc Natl Acad Sci U S A. 2018;115:5247–5252.
Yu L, Sawle AD, Wynn J, et al. Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum Mol Genet. 2015;24:4764–4773.
Wynn J, Aspelund G, Zygmunt A, et al. Developmental outcomes of children with congenital diaphragmatic hernia: a multicenter prospective study. J Pediatr Surg. 2013;48:1995–2004.
Wynn J, Krishnan U, Aspelund G, et al. Outcomes of congenital diaphragmatic hernia in the modern era of management. J Pediatr. 2013;163:114–119.
Homsy J, Zaidi S, Shen Y, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266.
Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601.
Farkouh-Karoleski C, Najaf T, Wynn J, et al. A definition of gentle ventilation in congenital diaphragmatic hernia: a survey of neonatologists and pediatric surgeons. J Perinat Med. 2017;45:1031–1038.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)-a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381.
Hollingshead A. Four factor index of social status. Yale J Sociol. 2011;8:21–51.
Bayley N. Bayley scales of infant and toddler development. 3rd ed. San Antonio, TX: Harcourt Assessments; 2006.
Harrison PL, Oakland T. Adaptive behavior assessment system. 2nd ed. San Antonio, TX: Harcourt Assessments; 2003.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84.
Chiang C, Layer RM, Faust GG, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12:966–968.
Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950.
Riggs ER, Church DM, Hanson K, et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet. 2012;81:403–412.
Werling DM, Brand H, An JY, et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet. 2018;50:727–736.
Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769.
Santosa F, Symes WW. Linear inversion of band-limited reflection seismograms. SIAM J Sci Stat Comput. 1986;7:1307–1330.
Tibshirani R. Regression shrinkage and selection via the Lasso. J R Stat Soc. 1996;58:267–288.
Longoni M, High FA, Qi H, et al. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum Genet. 2017;136:679–691.
Mous DS, Kool HM, Wijnen R, Tibboel D, Rottier RJ. Pulmonary vascular development in congenital diaphragmatic hernia. Eur Respir Rev. 2018;27:170104.
Sluiter I, van der Horst I, van der Voorn P, et al. Premature differentiation of vascular smooth muscle cells in human congenital diaphragmatic hernia. Exp Mol Pathol. 2013;94:195–202.
Acknowledgements
We thank the patients and their families for their generous contribution. We are grateful for the technical assistance provided by Patricia Lanzano, Jiangyuan Hu, Jiancheng Guo, Liyong Deng, Donna Garey, and Anketil Abreu from Columbia University. We thank our clinical coordinators and developmental specialists across the DHREAMS centers: Karen Lukas, Desiree White, and Jessica Conway at Washington University School of Medicine; Patricia Burns, Melissa Spare, and Sarah (Lexie) Price at Cincinnati Children’s Hospital; Sheila Horak and Kelly Miller at Children’s Hospital & Medical Center of Omaha; Jeannie Kreutzman and Jennifer Butcher at CS Mott Children’s Hospital; Tracy Perry at Monroe Carell Jr. Children’s Hospital; Michelle Kallis at Northwell Health, Brandy Gonzales, and Alicia McIntire at Oregon Health and Science University; Gentry Wools and Lorrie Burkhalter at Children’s Medical Center Dallas; Elizabeth Jehle at Hassenfeld Children’s Hospital at NYU Langone Health; Michelle Knezevich and Cheryl Kornberg at Medical College of Wisconsin; and Min Shi at Children’s Hospital of Pittsburgh. The genome sequencing data were generated through National Institutes of Health (NIH) Gabriella Miller Kids First Pediatric Research Program (X01HL132366, X01HL136998, and X01HL140543) and the University of Washington Center for Mendelian Genomics with National Human Genome Research Institute (NHGRI) grant HG006493. This work was supported by NIH grants R01HD057036 (L.Y., J.W., W.K.C.), R03HL138352 (W.K.C., Y.S.), R01GM120609 (Y.S.), UL1 RR024156 (W.K.C.), and 1P01HD068250 (W.K.C., Y.S.). Additional funding support was provided by grants from CHERUBS, CDHUK, and the National Greek Orthodox Ladies Philoptochos Society, Inc. and generous donations from Fore Hadley, the Williams Family, Wheeler Foundation, Vanech Family Foundation, Larsen Family, Wilke Family, and many other families. S.N. received salary support through a Ruth L. Kirschstein National Research Service Award of the NIH under award number 5T32HL007854-22. The genome sequencing data can be obtained from dbGAP through accession phs001110.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Qiao, L., Wynn, J., Yu, L. et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet Med 22, 2020–2028 (2020). https://doi.org/10.1038/s41436-020-0908-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0908-0
Keywords
This article is cited by
-
Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays
European Journal of Human Genetics (2023)
-
Congenital diaphragmatic hernia
Nature Reviews Disease Primers (2022)
-
Neuropsychological outcome in survivors of congenital diaphragmatic hernia at 5 years of age, what does it tell?
European Journal of Pediatrics (2022)
