
Abstract
Purpose
Plexiform neurofibromas (pNF) develop in children with neurofibromatosis type 1 (NF1) and can be associated with several skeletal comorbidities. Preclinical mouse studies revealed Nf1 deficiency in osteoprogenitor cells disrupts, in a MEK-dependent manner, pyrophosphate (PPi) homeostasis and skeletal mineralization. The etiology of NF-associated skeletal manifestations remains unknown.
Methods
We used mouse models of NF1 neurofibromas to assess bone mineralization of skeletal structures adjacent to tumors. Expression of genes involved in pyrophosphate homeostasis was assessed in mouse and human NF tumors and Schwann cell cultures. We used dual-energy X-ray absorptiometry (DXA) to assess tumor-associated changes in bone mineral density (BMD) in an individual with NF1 following treatment with the MEK inhibitor selumetinib.
Results
We detected increased nonmineralized bone surfaces adjacent to tumors in mouse models of NF1 neurofibromas. Expression of Enpp1, a PPi-generating ectophosphatase, and ANKH, a PPi transporter, was increased in mouse and human neurofibroma-derived tissues and Schwann cells, respectively. In one patient, tumor-associated reductions in BMD were partially rescued following therapy with selumetinib.
Conclusion
Results indicate that NF-associated skeletal pathologies in NF1 are associated with dysregulated pyrophosphate homeostasis in adjacent NF tumors and suggest that treatment of NFs with MEK inhibitors may improve skeletal manifestations of the disease.
Similar content being viewed by others

INTRODUCTION
Neurofibromatosis type 1 (NF1) is a cancer-predisposing genetic syndrome caused by inherited or sporadic variants in the NF1 gene.1 Twenty to 50 percent of individuals with NF1 present with plexiform neurofibromas (NF),2,3 which are congenital tumors that grow over the lifetime and are thus more apparent in older individuals. These Schwann cell–derived tumors enlarge most prominently during the first decade of life4,5 and, like many other NF1 manifestations, arise from somatic variants of the wild-type (WT) NF1 allele or from loss of NF1 heterozygosity. Loss of neurofibromin, encoded by NF1, allows for persistent RAS activity and thereby leads to activation of the RAS/MEK/ERK pathway. Recently, the MEK inhibitor selumetinib was shown to significantly reduce NF burden in children with NF1.6,7
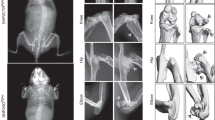
As many as 30% of individuals with NF1 develop skeletal manifestations, including osteopenia, tibial dysplasia and pseudarthrosis, or scoliosis.8 While somatic loss of NF1 in bone cells was detected in tibial pseudarthrosis9,10,11 and dystrophic scoliosis cases,12 and while the different types of spinal deformities (with dystrophic features or not) in NF1 are well described,13,14,15 NF-associated skeletal manifestations have been less investigated. Individuals with NF1 presenting with paraspinal neurofibromas have sixfold higher odds of developing scoliosis compared with patients without paraspinal tumors, and up to 63% of NF1 individuals with dystrophic scoliosis have evidence of paraspinal tumors on magnetic resonance imaging (MRI).16,17,18 Although “erosive” defect of bone from contiguous neurogenic tumors was suspected, no direct evidence beyond X-rays and bone mineral density (BMD) are available to explain how tumors may alter adjacent skeletal elements.19,20 Figure 1a, b illustrates one such patient with rapidly progressing dystrophic scoliosis associated with vertebral body and rib deformities immediately adjacent to a plexiform neurofibroma. Plexiform tumors affecting the extremities may also present with progressive skeletal deformity, bone loss, and length discrepancy. Figure 1c, d depicts a patient with unilateral leg NF and progressive deformity of the pelvis, acetabulum, and proximal femur associated with the tumor. Subtrochanteric femoral diameter of the tumor-laden leg in this patient decreased over 7 years (Fig. 1e). Thus, skeletal comorbidities exist at multiple sites among individuals with NF1 and NFs.

(a) Standing X-ray (top) and magnetic resonance image (MRI) (bottom) of an adolescent female patient with NF1 NF-associated dystrophic scoliosis. The scoliotic curve progressed from a Cobb angle of 40° to 79° in one year. (b) Three-dimensional computed tomography (CT) spine reconstructions of the same patient. White arrows indicate vertebral body (top) and rib head (bottom) deformity on the concave side adjacent to tumor that are not evident on the convex side or further from the tumor (black arrows). (c) Longitudinal X-ray imaging of a child with bone loss associated with a unilateral NF extending from the left pelvis throughout the entire extremity. White arrows show deformity of the pelvis, acetabulum, and proximal femur. (d) MRI imaging at presentation depicts the location and involvement of the NF. (e) Quantification of tumor-associated femur deformity by serial measurements of the subtrochanteric femur diameter. Affected: tumor-laden femur; unaffected: contralateral side. X-rays with measurement sites are shown in Supplementary Fig. 2.
Results from conditional knock-out (cKO) mouse models of Nf1 deletion in the bone mesenchymal lineage and expression profiling of patient-derived bone stromal cells supported a model whereby loss of NF1 leads to activation of genes promoting the accumulation of inorganic pyrophosphate (PPi), a strong inhibitor of hydroxyapatite formation and bone mineralization.21 We have shown that upregulation of PPi pathway–related genes in mouse osteoprogenitor cells following deletion of Nf1, including Enpp1 (an ectophosphatase generating PPi), Ank (a channel transporting PPi in the extracellular milieu), and Spp1 (Osteopontin, an extracellular protein inhibiting hydroxyapatite formation), is MEK-dependent.21 In this study, we investigated whether the expression of genes involved in PPi homeostasis was altered in NFs and whether skeletal changes were associated with adjacent neurofibromas. Furthermore, we present an individual with NF1 presenting with reduced BMD associated with a unilateral NF who saw marked improvement in BMD concomitant with response of the tumor to MEKi therapy.
MATERIALS AND METHODS
Mouse models
Control and tamoxifen-inducible Plp-creERT; Nf1flox/flox mice22,23,24 received tamoxifen (75 mg/kg) intraperitoneally (i.p.) twice daily for 3 days at 2 months of age. Periostin-cre; Nf1flox/flox mice24,25,26 and Dhh-cre; Nf1flox/flox mice24,27 were generated as described previously. No randomization was used for animal studies. Mice were euthanized between 8 and 10 months of age. Both genders were included in the measurements. All mouse procedures were approved by the Institutional Animal Care and Use Committee (IACUC) boards at Indiana School of Medicine and NCI.
Histological measurements
Tissues were immersion-fixed in PBS-based 4% paraformaldehyde and embedded undecalcified in methyl methacrylate before staining by von Kossa/van Gieson to stain mineralized bone and nonmineralized collagenous bone matrix, respectively. Histomorphometric measurements were performed using the Bioquant Image Analysis System (R&M Biometrics, Nashville, TN), blinded to genotype and following American Society for Bone and Mineral Research (ASBMR) guidelines.28,29 Osteoid parameters were measured directly by manual tracing from images taken at 20× magnification.
Immunocytochemistry
Sections were deplastified before rehydration, followed by immunohistochemistry (IHC) using an antibody against S100 (clone 15E2E2, Sigma, MAB079–1), a HRP-coupled goat antimouse IgG (Abcam, ab6789), and the Dako Liquid DAB + Substrate Chromogen System (Agilent Technologies, K346811–2).
Human Schwann cell gene expression analysis
Enrichment and culture of NF1−/− Schwann cells from NFs was done as described previously.30 Normal human embryonic Schwann cells (ScienCell Research Laboratories, Carlsbad, CA) were cultured as NF1−/− Schwann cells. RNA-Seq libraries were constructed from 1 µg total RNA after ribosomal RNA depletion (Ribo-Zero GOLD and Illumina TruSeq RNA Sample Prep V2 Kit, Illumina, San Diego, CA). Data were processed using RTA v.1.18.64 and CASAVA v.1.8.2. software. Data for cultured Schwann cells from NFs of individuals with NF1 (N = 23) and cultured normal human embryonic Schwann cell controls (N = 7) were further processed using standard Tuxedo pipeline.31 Resulting gene expression data sets were then Log2 transformed. Genes with median Log2 FPKM score below 0.5 in both groups were considered not expressed and removed from further analysis.
Human and mouse gene expression microarray data
We reanalyzed published human and mouse (GEO accession GSE14038, GSE41747) microarray data sets, including human nerve, human NF tumor, mouse nerve, and mouse neurofibroma samples.32,33 Raw CEL files were preprocessed and gene expression levels were normalized (Bioconductor/affy package’s Robust Multiarray Average method).34 Differentially expressed genes were predicted using Bioconductor/limma package.35
RNASeq analysis
RNA was isolated from trigeminal nerve tissues from Periostin-cre; Nf1flox/flox mice (RNeasy Plus Mini Kit, Qiagen) with DNase I treatment (Qiagen) for 15 minutes. Then, 400 ng total RNA was used as input for RNAseq library construction (Kapa Stranded mRNA kit). TruSeq adapter sequences were used for indexing. Library was polymerase chain reaction (PCR)-amplified for 12 cycles. Equimolar amounts of each library were run as 12-plexed 1.65 pM pools, single-indexed, on a NextSeq 500–75 cycle, high output V2 kit to produce single-end reads. Quality control (QC)-passed reads were aligned to the mouse reference genome (mm10) using STAR 2.4.2a36 and reads were translated to transcriptome coordinates using Salmon 0.60.37 Isoform data were collated to single-gene IDs using the R package biomaRt,38 and abundance estimates were upper quartile normalized using R. Abundance values for genes were used as input for DESeq2 to identify differentially expressed genes39 using an adjusted p value cutoff of < 0.05.
Whole-body dual-energy X-ray analysis
Children aged 5–16 enrolled at the National Institutes of Health (NIH) on a phase 2 clinical trial of selumetinib (SPRINT, NCT01362803) underwent a baseline dual-energy X-ray analysis (DXA) of whole body, proximal femur, and lumbar spine using a Hologic Discovery A, using standard positioning technique. Patients found to have an abnormal result in any of these body regions (Z-score ≤ −2.0) had repeat imaging performed after 1 year on treatment. Height and age-adjusted Z-scores were calculated for whole body, total body less head (a.k.a. “subtotal”), lumbar spine, total hip, and femoral neck using data from the Bone Mineral Density in Childhood Study. One subject out of 11 was excluded due to hip surgery during the study period.
Femoral diameter measurements
Femur diameter was measured directly from X-ray films taken as standard-of-care. Locations of diameter measurement are shown in Supplementary Fig. 2.
Statistics
All gene expression data and distributions are represented as boxplots. Pair-wise comparisons were performed using standard two-sided t-tests in R.
Ethics statement
The study was approved by the institutional review board at the National Cancer Institute, and all participants provided written informed consent.
RESULTS
We first evaluated differences in skeletal mineralization adjacent to Nf1-deficient neurofibroma tumors using the inducible Plp-CreERT; Nf1flox/flox cKO mouse model, where the Nf1flox allele is conditionally deleted in myelinating Schwann cells at 2 months of age under the control of the Myelin proteolipid protein (Plp) promoter.23 Histological examination of vertebral bones from mutant mice euthanized at 9–11 months of age, at a time where large paraspinal plexiform neurofibromas associated with peripheral nerves and dorsal root ganglia are readily detectable, showed vertebral processes surrounded by S100+ NFs that appeared completely unmineralized (Supplementary Fig. 1a). We also observed cases of tumors infiltrating the pelvic bones, where nonmineralized bone matrix (osteoid) and S100+ tumors were colocalized (Supplementary Fig. 1b). Trabecular osteoid surfaces and volumes were increased in the vertebrae of Plp-creERT; Nf1flox/flox mice adjacent to S100+ neurofibromas, although to a lesser extent than in vertebral processes immediately adjacent to tumors. In mutant mice, this increase in cancellous osteoid parameters was not evident in vertebrae adjacent to normal nerves (Fig. 2a, b) and bone volume over tissue volume (BV/TV) in lumbar vertebrae not associated with tumors was normal (BV/TV: 18.06 ± 5.8 in WT versus 18.30 ± 4.8 in cKO, n = 6–9). In a second mouse model in which the Cre recombinase is driven by the Desert Hedgehog (Dhh) promoter to conditionally ablate the Nf1flox allele in embryonic Schwann cell precursors (Dhh-cre; Nf1flox/flox),27 increased vertebral osteoid volume and surface adjacent to neurofibroma tumors were also observed (Supplementary Fig. 1c). These findings demonstrate, in two independent Schwann cell–specific Nf1 conditional knockout mouse models, the existence of altered mineralization closely associated with neurofibromas.

(a) Representative images of von Kossa/van Gieson-stained (top) and S100+ nerves (bottom) thoracic vertebral sections from control and Plp-CreERT; Nf1flox/flox mice. N: normal nerve, T: plexiform tumor adjacent to vertebral bone. Osteoid (nonmineralized bone) is stained pink (white arrows) and mineralized bone is stained black. Bar: 50 µm. (b) Osteoid surfaces/bone surfaces (OS/BS), osteoid volume/bone volume (OV/BV) and osteoid width (O. Wi.) in vertebral cancellous bone regions adjacent to normal nerves or neurofibroma (NF) tumors in control and Plp-creERT;Nf1flox/flox (cKO) mice (wild type [WT]: n = 6 vertebral bodies from 3 mice), cKO without adjacent tumor (−, n = 9 vertebral bodies from 4 mice) and adjacent to tumor (+, n = 7 vertebral bodies from 4 mice). *p < 0.05, one-way analysis of variance (ANOVA) and post hoc pair-wise comparison with Bonferroni adjustment. (c, d) Normalized expression levels in neurofibromas from (c) Dhh-cre;Nf1flox/flox (n = 15) and control (normal sciatic nerve, n = 15) mice (microarray) and (d) from Periostin-cre; Nf1flox/flox (n = 6) and control (trigeminal nerve, n = 6) mice (RNAseq). *p < 0.05; **p < 0.01 by two-sided t-test.
To determine whether dysregulation of PPi homeostasis potentially contributed to neurofibroma-associated mineralization defects, and because the measurement of tissue PPi level was technically not feasible, we measured expression of genes promoting PPi generation and extracellular transport in tumors from multiple mouse models. Microarray analysis of neurofibromas from Dhh-cre; Nf1flox/flox mice compared with WT sciatic nerves (GSE41747)32 identified increased expression of Enpp1 in neurofibroma tumors, whereas expression of Ank was reduced (Fig. 2c). Transcriptome profiling of trigeminal nerve tumors from Periostin-cre; Nf1flox/flox mice25 and control nerves confirmed these results in an independent mouse model of Nf1-deficient neurofibromas (Fig. 2d), in which Cre-mediated Nf1 ablation occurs in the neural crest-–derived Schwann cell lineage after embryonic day E11.
We then tested whether activation of the PPi pathway was also evident in human NF1 NFs. Transcriptome profiling of low-passage Schwann cell cultures from NFs of individuals with NF1 showed increased expression of ANKH compared with embryonic Schwann cells (Fig. 3a), but no difference in ENPP1 expression (not shown). Independent microarray expression analysis of human NF tissues (GSE14038, GSE41747)33 confirmed higher ANKH expression compared with normal sciatic nerves (Fig. 3b). Although not statistically significant, expression of ENPP1 trended higher in NF1 NFs compared with control (Fig. 3b).

(a) Gene expression in cultured Schwann cells (SCs) from NFs of individuals with neurofibromatosis type 1 (NF1) (n = 24) versus cultured embryonic Schwann cell controls (n = 7) (RNAseq). (b, c) Gene expression in human NF1 NF tissue (n = 13) versus normal sciatic nerve control tissue (n = 3) (microarray). *p < 0.05; ***p < 0.001 by two-sided t-test.
Results presented here from both conditional mouse models and patient-derived tumors concur to support the notion that activation of genes regulating PPi homeostasis in NF1 neurofibromas is associated with decreased bone mineralization and likely contributes to skeletal comorbidities in children with NF1. The response of NF1 NFs to MEK inhibitor (MEKi) therapy6 and the MEK dependency of PPi pathway activation21 thus implied that treatment of NF1 plexiform tumors with MEK inhibitors may have beneficial effects on bone mineralization. To investigate this model further, baseline and post-therapy whole-body DXA scans of ten subjects from the National Cancer Institute phase 1/2 clinical trial (NCT01362803) of selumetinib for inoperable NFs were examined. Of these, one subject had a unilateral NF of the leg that allowed for comparison of BMD to the contralateral leg without tumor. In this patient, BMD of the tumor-laden leg was dramatically reduced compared with the contralateral leg without tumor; this difference was not observed among participants without leg NF (Fig. 4a, baseline). Following 1 year of MEKi therapy, the subject’s NF showed a partial response with volumetric reduction of 23% (1267 mL vs. 973 mL), and the difference in BMD between tumor-laden and contralateral leg improved from −15% to −8% (Fig. 4a, post-MEKi). Whereas 1-year of MEKi therapy did not affect total hip and femoral neck adjusted Z-scores in subjects without leg NFs, it markedly improved these parameters in the affected leg of this patient (Fig. 4b, c).

(a) Difference in BMD between left and right legs is shown for 9 individuals without leg neurofibroma (NF) (gray circles) and one patient with a left-leg NF (blue diamond) at baseline (left) and following 1-year MEKi therapy (right). Magnetic resonance image (MRI) shows left-sided tumor involvement and volumetric reduction following therapy. (b, c) Height adjusted total hip (b) and femoral neck (c) Z-scores at baseline and following 1-year MEKi therapy.
DISCUSSION
MEK-dependent PPi pathway activation in NFs suggests tumor-associated skeletal comorbidities in NF1 result, at least in part, from a local inhibition of bone mineralization. The rate of bone accrual and mineralization is high during childhood/adolescence and coincides with the initial active growth of NFs and, importantly, their response to MEKi therapy. Therefore, our findings highlight a common therapeutic window where MEKi therapy may reduce both tumor burden and tumor-associated PPi pathway activation, allowing for proper mineralization of the affected bony structures in children. This secondary therapeutic effect was observed in the neurofibroma-laden femur/hip of a single available patient with unilateral neurofibroma, despite the short-term duration of the treatment and the remaining presence of tumor post-treatment. This effect was not observed in the skeleton of patients without adjacent tumors, thus supporting it is specific to the skeleton associated with regional PN. Because of the preliminary nature of our findings in a single human patient, longitudinal evaluation of additional patients will be necessary to confirm efficacy of MEK inhibitors to treat the skeletal manifestations associated with neurofibroma tumors, and to address potential effects at other bone sites and on bone growth. This PPi-dependent mechanism will also require confirmation of increased PPi generation in plexiform tumors and does not exclude other mechanisms such as alterations in blood flow or nutrient utilization.
The model proposed here may apply to other neurofibroma-associated skeletal manifestations of NF1, including craniofacial abnormalities associated with orbital NFs, dystrophic scoliosis, and, potentially, any skeletal structure adjacent to NFs. The potential clinical utility of MEKi may thus extend beyond the treatment of NFs to positively impact the many neurofibroma-associated skeletal comorbidities in children with NF1, thus supporting the need for integrated, comprehensive, and collaborative care clinics for patients with NF1. Lastly and importantly, results from this study highlight the opportunity for ongoing and future clinical trials testing MEK inhibitors as therapeutics for NF1 NFs to investigate skeletal comorbidities, which may inform the design of more effective clinical management strategies for patients with NF1.
References
Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834–843.
Mautner VF, Asuagbor FA, Dombi E, et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008;10:593–598.
Plotkin SR, Bredella MA, Cai W, et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012;7:e35711.
Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007;68:643–647.
Nguyen R, Dombi E, Widemann BC, et al. Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis. 2012;7:75.
Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375:2550–2560.
Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382:1430–1442.
Elefteriou F, Kolanczyk M, Schindeler A, et al. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A. 2009;149A:2327–2338.
Stevenson DA, Zhou H, Ashrafi S, et al. Double inactivation of NF1 in tibial pseudarthrosis. Am J Hum Genet. 2006;79:143–148.
Paria N, Cho TJ, Choi IH, et al. Neurofibromin deficiency-associated transcriptional dysregulation suggests a novel therapy for tibial pseudoarthrosis in NF1. J Bone Miner Res. 2014;29:2636–2642.
Sant DW, Margraf RL, Stevenson DA, et al. Evaluation of somatic mutations in tibial pseudarthrosis samples in neurofibromatosis type 1. J Med Genet. 2015;52:256–261.
Margraf RL, VanSant-Webb C, Mao R, et al. NF1 somatic mutation in dystrophic scoliosis. J Mol Neurosci. 2019;68:11–18.
Crawford AH. Pitfalls of spinal deformities associated with neurofibromatosis in children. Clin Orthop Relat Res. 1989;245:29–42.
Durrani AA, Crawford AH, Chouhdry SN, Saifuddin A, Morley TR. Modulation of spinal deformities in patients with neurofibromatosis type 1. Spine. 2000;25:69–75.
Ramachandran M, Tsirikos AI, Lee J, Saifuddin A. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004;17:483–491.
Lykissas MG, Schorry EK, Crawford AH, Gaines S, Rieley M, Jain VV. Does the presence of dystrophic features in patients with type 1 neurofibromatosis and spinal deformities increase the risk of surgery? Spine. 2013;38:1595–1601.
Nguyen R, Dombi E, Akshintala S, Baldwin A, Widemann BC. Characterization of spinal findings in children and adults with neurofibromatosis type 1 enrolled in a natural history study using magnetic resonance imaging. J Neurooncol. 2015;121:209–215.
Hu Z, Liu Z, Qiu Y, Xu L, Yan H, Zhu Z. Morphological differences in the vertebrae of scoliosis secondary to neurofibromatosis type 1 with and without paraspinal neurofibromas. Spine. 2016;41:598–602.
Hunt JC, Pugh DG. Skeletal lesions in neurofibromatosis. Radiology. 1961;76:1–20.
Lodish MB, Dagalakis U, Sinaii N, et al. Bone mineral density in children and young adults with neurofibromatosis type 1. Endocr Relat Cancer. 2012;19:817–825.
de la Croix Ndong J, Makowski AJ, Uppuganti S, et al. Asfotase-alpha improves bone growth, mineralization and strength in mouse models of neurofibromatosis type-1. Nat Med. 2014;20:904–910.
Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72.
Mayes DA, Rizvi TA, Cancelas JA, et al. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res. 2011;71:4675–4685.
Zhu Y, Romero MI, Ghosh P, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876.
Rhodes SD, He Y, Smith A, et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum Mol Genet. 2019;28:2752–2762.
Lindsley A, Snider P, Zhou H, et al. Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Dev Biol. 2007;307:340–355.
Wu J, Williams JP, Rizvi TA, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116.
Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Committee. J Bone Miner Res. 1987;6:595–610.
Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2–17.
Muir D, Neubauer D, Lim IT, Yachnis AT, Wallace MR. Tumorigenic properties of neurofibromin-deficient neurofibroma Schwann cells. Am J Pathol. 2001;158:501–513.
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578.
Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–347.
Miller SJ, Jessen WJ, Mehta T, et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol Med. 2009;1:236–248.
Gautier L, Cope L, Bolstad BM, Irizarry RA. affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315.
Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419.
Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Acknowledgements
The authors thank Gerard Portela, Mark Ahlman, John Shern, Stuart Almond, B. Stephens Richards, and Molly Dempsey for their help with different aspects of this work. Research reported in this publication was supported by the Department of Defense (NF140019, F.E.), the Texas Neurofibromatosis Foundation (J.J.R), and Texas Scottish Rite Hospital for Children (J.J.R.). F.E. (R21-AR072483, R01-AG055394), N.R. (R01-NS28840), J.J.R and D.W.C. (U54-CA196519-04), and S.D.R. (K12-HD000850) were supported by funding from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
F.E. received research funding from Alexion (not related to this paper). The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ma, Y., Gross, A.M., Dombi, E. et al. A molecular basis for neurofibroma-associated skeletal manifestations in NF1. Genet Med 22, 1786–1793 (2020). https://doi.org/10.1038/s41436-020-0885-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0885-3
Keywords
This article is cited by
-
From Stem to Sternum: The Role of Shp2 in the Skeleton
Calcified Tissue International (2022)
-
MEK inhibitors - novel targeted therapies of neurofibromatosis associated benign and malignant lesions
Biomarker Research (2021)
