
Abstract
The role of X-inactivation is often ignored as a prime cause of sex differences in disease. Yet, the way males and females express their X-linked genes has a major role in the dissimilar phenotypes that underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the heart, blood, skin, muscle, and bones. Summarized here are many examples of the different presentations in males and females. Other data include reasons why women are often protected from the deleterious variants carried on their X chromosome, and the factors that render women susceptible in some instances.
Similar content being viewed by others
INTRODUCTION
Sex differences in human disease are usually attributed to sex specific life experiences, and sex hormones that influence the function of susceptible genes throughout the genome.1,2,3,4,5 Such factors do account for some dissimilarities. However, a major cause of sex-determined expression of disease has to do with differences in how males and females transcribe their gene-rich human X chromosomes, which is often underappreciated as a cause of sex differences in disease.6 Males are the usual ones affected by X-linked pathogenic variants.6 Females are biologically superior; a female usually has no disease, or much less severe disease than the male with the same variant, unless she is homozygous for the deleterious allele, or it is lethal for males.
The X chromosome carries 867 known protein coding genes.7 Clearly, pathogenic variants that induce complete loss of function may be lethal to fetuses of both sexes; however, a number of these pathogenic variants—less severe, or occurring in less-essential genes—cause at least 533 X-linked diseases8 that affect males more severely.8 Rather than influencing sexual development, most of these genes play a role in nonreproductive human tissues, including brain, bone, blood, ears, heart, liver, kidney, retina, skin, and teeth.
Table 1 provides data about a substantial number of X-linked disorders obtained in large part from OMIM8 that confirm the lesser susceptibility of females. The table is not all-inclusive, but it provides enough data to show the greater severity of these diseases in males, and to illustrate why some, but not all, females with the same X-linked deleterious allele are protected from its effects. This paper is motivated by the question: When so many women are protected from manifesting severe X-linked diseases, why are some of them susceptible?
SEX DIFFERENCES ARE DUE TO X-INACTIVATION
The sex differences in the effect of X-linked pathologic variants is due to our method of X chromosome dosage compensation, called X-inactivation;9 humans and most placental mammals compensate for the sex difference in number of X chromosomes (that is, XX females versus XY males) by transcribing only one of the two female X chromosomes. X-inactivation silences all X chromosomes but one; therefore, both males and females have a single active X.10,11
For 46 XY males, that X is the only one they have; it always comes from their mother, as fathers contribute their Y chromosome. However, because X chromosomes are silenced in a random fashion, females usually have two kinds of cells in every tissue; those with their maternal X active and those with an active paternal X. Females are protected to a large extent because their two X chromosomes most often differ in genetic content.
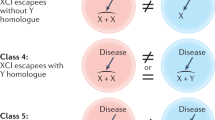
Sex differences in diseases due to deleterious variants encoded by the X chromosome originate from the sex difference in the expression of the variant allele; if present in male tissues, it is expressed in every cell, but if present in female tissues, the variant is usually expressed in only half the cells (Fig. 1).

Adapted from Fig. 1 of Franco B, Ballabio A. Curr Opin Genet Dev. 2006;16:254–259, with permission of authors. In females: On average, the ratio of the two cell types (expressing normal or variant alleles) is approximately 50:50. However, the ratio may differ because of chance, or a selective disadvantage for cells expressing the variant. Extreme divergence from the 50:50 ratio, known as skewing of XCI, may differ from tissue to tissue, and among individuals influencing the severity of the phenotype. Cell selection usually takes place only in cells which express the variant and which do not receive the essential gene products from the normal cells that make them. In males: an X-linked variant is expressed in every cell. The exceptions are males with somatic mosaicism: like females, mosaic males will express both variant and normal allele, and the phenotype depends on the admixture of variant and normal cells.
FEMALES ARE MOSAICS
A woman is less susceptible to the pathogenic variants in genes on her active X chromosome because the variant is not expressed in all her cells.12
FEMALES CAN AMELIORATE THE EFFECTS OF PATHOGENIC VARIANTS
Most women do not manifest X-linked disorders because (1) they are not homozygous for the pathogenic variant, and (2) their variant cells (those expressing the deleterious allele) receive sufficient gene product to carry out the essential metabolic function from the cells that transcribe the normal allele.
The crucial protein is provided in one of two ways. Either the cells that can make it transfer it to the deficient cells, or, if this cell-to-cell transport does not take place, the lack of functional protein may cause the deficient cells to divide less efficiently, and so they are eventually overgrown by the cells that make the normal protein. Yet, in various tissues, cells differ in their capacity to transfer gene products and so there may be differences within body tissues in the ability of the normal cell to share, or the abnormal cell to survive12 (Fig. 1, Table 1).
FEMALES ALMOST ALWAYS HAVE LESS SEVERE MANIFESTATIONS OF X-LINKED DISEASES
For most X-linked deleterious variants, the manifestations are less severe in females than males. The mix of normal and abnormal cells moderates females' disease. Yet, there are disorders in which the variant is so lethal that most males with severe deficiency of the gene product die in utero. Because the only survivors are females or mosaic males, who also have a mix of variant and normal cells, they are the ones with nonlethal manifestation of the disorder. X-linked diseases, such as incontinentia pigmenti, or orofacial digital syndrome type 1, occur only in females or mosaic males.13
SOME FEMALES ARE MANIFESTING HETEROZYGOTES
Many factors determine if a heterozygote manifests her mosaic variant. Our knowledge of the effect of second variants of genes in related pathways on the ultimate phenotype is meager, but suggests that we need to be aware of the potential effect of other relevant genes on any variant. Although Table 1 includes only one known instance of digenic X variants (Dent disease 2), one expects there will be more. The effect of digenic variants leading to Dent disease are ameliorated in the heterozygote, who carries both variants on the same X chromosome, as these abnormal cells are strongly disfavored (Table 1).8 In this case, heterozygotes are much less severely affected than hemizygous males because of the strong cell selection against cells expressing the two variants simultaneously.
EFFECT OF SEX AND X-INACTIVATION ON PHENOTYPE IN X-LINKED DISORDERS
As I learn about why some heterozygotes express their variants, yet others with the same variant do not, I more fully appreciate the nuances of disease processes and the intricacy of the interdependence between the type of cell, its response to abnormal gene products, and the nature of the disease variant. Table 1 shows the effect of pathogenic variants of X-linked genes in males and females. For almost all of the disorders listed, females have less severe clinical manifestations. Table 1 also documents the variables that determine if an X chromosomal variant is manifested or not, that is, if products are shared or if normal cells have a selective advantage. The severity of the phenotype is most often attributable to the nature of the variant. Does it completely eliminate the gene’s function, or does it allow some gene product to be produced? Variants that disable an essential protein completely are more often seen in females, as males whose only gene is nonfunctional are lost in utero, unless other genes can substitute for it. On the other hand, variants with some residual function may affect only males. In both cases, the female phenotype is less severe, as she has some normal cells that carry out the essential function.
EFFECT OF SHARING GENE PRODUCT BETWEEN CELLS
Because it is largely unknown, Table 1 does not include the quantity of gene product that must be provided to a cell to replace that lost through a variant. In most cases 50% activity is more than enough, and for many genes, even smaller amounts of product will suffice. This readily explains why many X-linked disorders never affect women. For example, less than 5% of the enzyme HPRT can alter the phenotype from severe hyperuricemia, seen in Lesch–Nyhan males, to gout. Lesch–Nyhan heterozygotes rarely express any features of the syndrome, including gout. In most of her tissues the product of the HPRT reaction, inosinic acid, is transferred from her cells that synthesize it, to those that cannot, through gap junctions, present in all cells of the body except blood cells.14
Cell sharing also occurs in women who are heterozygous for variants causig X-linked lysosomal diseases. Lysosomes contain many enzymes that break down proteins and lipids. Variants in the genes that encode these enzymes cause diseases by the accumulation of undegraded material within the lysosomes of affected individuals. Fortunately, the deficient cells of the heterozygote can take up the enzyme secreted by the normal cells through a process called endocytosis. Therefore, potential manifestations in carriers of variants of lysosomal enzymes encoded by the X chromosome are generally ameliorated by the transfer of these enzymes from the cells that can make them to those that cannot.15
EFFECT OF CELL SELECTION IN MOSAIC FEMALES
Because they lack gap junctions, the leukocytes from Lesch–Nyhan heterozygotes do not receive the inosinic acid made by their normal counterparts. Fortunately, the lack of inosinic acid slows the rate of cell division; the normal cells (because the variant is on their inactive X) eventually replace the deficient ones. As a consequence, heterozygous mothers and sisters of Lesch–Nyhan males have only normal blood cells, and in other tissues, their mutant cells share the gene products provided by their normal cells.16 It takes about ten years for all their abnormal red cells to be replaced17 because in this case, the selective advantage of the normal allele is relatively small.
The rate of loss of deficient cells can be slow or fast depending on the degree to which the variant is disadvantageous. When selection is intense, that is, when it severely disfavors the cells that express the variant, heterozygous females benefit, as they rapidly lose all their mutant cells. Sometimes the loss of variant cells occurs so early that abnormal cells may never enter a tissue. In cases of immunodeficiencies, like Wiskott–Aldrich syndrome, the growth disadvantage is immediate; the mutant B cell precursors never leave the bone marrow (Table 1).8
Unfortunately, sometimes it is the variant cell that has the selective advantage. For reasons not yet understood, heterozygotes with the variant causing adrenoleukodystrophy slowly lose their population of wild type cells.18 Therefore, as they age, they usually manifest some symptoms of the disease. Adrenoleukodystrophy is the only known X-linked disease where the variant allele has a selective advantage. X-linked variants that slow the growth of the expressing cells usually protect females from manifesting them. If one examines the cells from these heterozygotes, one sees highly skewed patterns of X-inactivation in the expressing tissues: eventually, the X with the variant will always be silent (that is, always on the inactive X). Females carrying pathologic variants that produce X-linked mental retardation often have X-inactivation patterns that are highly skewed favoring expression of the wild type gene, protecting them from ill effects of their variant.19 In such women, the cells that express the variant gene are disfavored.
MANIFESTING HETEROZYGOTES
With so many mechanisms available to protect females with an X-linked pathologic variant, one wonders why some heterozygotes manifest any symptoms of a disease. As mentioned previously, if the variant is so functionless that it is lethal to fetal or newborn males, then females with a single mutant allele are the only ones that can have the disease. As Table 1 indicates, the severity of the variant can determine if females express the variant or not.
Occasionally, females are the ones to manifest a disease because the variant allele interacts with the normal one, undergoing a process called cellular interference. The most well defined example of this is the craniofrontonasal syndrome, caused by a deficiency of Ephrin-B1 (EFNB1).20 Other members of the ephrin family of proteins can substitute for a deficiency of EFNB1 in males with the deleterious variant, and as a result, they have minimal clinical symptoms. Heterozygotes, however, have a mixture of mutant and wild type cells; for reasons not yet understood, it seems that such mixtures do not permit ephrin substitutes, and consequently, females have a deficiency more severe than that in males, who can substitute one ephrin for another. It is heterozygosity, not the complete loss of function, that produces the severe disorder. It is the patchwork or mosaic loss of EFNB1 that disturbs tissue boundary formation at the developing coronal suture. Several forms of infantile epilepsy also show similar cellular interference, but fortunately no other examples are known.
Yet, there are females who express an X-linked pathogenic variant, even though most carriers of pathogenic variants in similar genes do not. For example, females with Fabry disease, caused by lack of the lysosomal enzyme ɑ-galactosidase may have some of the clinical symptoms seen in affected males, whereas carriers of a variant in a gene encoding another lysosomal enzyme, iduronic sulfatase, rarely have any clinical symptoms associated with Hunter syndrome.21 Both enzymes are made in the lysosomes, and can be transported from the lysosomes of one cell to those of another cell. The two lysosomal disorders differ because iduronate sulfatase is taken up by cells better than the low uptake enzyme, ɑ-galactosidase.22,23 This difference in the ability of the lysosome to take up a product is responsible for normal Hunter heterozygotes and manifesting Fabry heterozygotes.
OTHER DETERMINANTS OF MANIFESTING HETEROZYGOTES
One wonders why some heterozygotes express disorders that do not affect most of the others with the same variant. Some females manifest their X-linked variant because it is overexpressed; their variant is expressed in more than half their cells because of skewing in the proportions of normal and abnormal cells. Although random inactivation usually means that 34–68% of heterozygous cells are abnormal,12 a few heterozygotes have more than 90% variant cells. Such females manifest the disorder because their wild type allele is not expressed sufficiently. These manifesting heterozygotes are often reported in medical journals because they are affected with a disease that usually occurs only in males. In some cases, a chance chromosome translocation or deletion is responsible for the skew, as such abnormal X chromosomes often influence the direction of skewing. X/autosome translocation chromosomes are responsible for the rare females afflicted with Duchenne muscular dystrophy.24 Maternal isodisomy,25 early onset monozygotic twinning (known to promote skewing26,27), and other extreme skewing of X-inactivation28 have also been responsible for manifesting heterozygotes (Table 2).
Although some skewing may be attributable to abnormalities of the XIST locus,29 such variants occur only rarely. If one XIST allele loses function, than that allele is always on the active X. Minor loss of function variants of XIST may lead to some skew,29 but few have been reported. Cell selection and random skewing are the most frequent causes of nonrandom X-inactivation. Random skewing frequently occurs because of events surrounding twinning, and confined placental mosaicism.30 Often skewing is due to the randomness of the inactivation process, which is stochastic and therefore due to chance. Ten percent of females are >2 standard deviations from the mean of 50%.23
Stochastic skewing that favors cells that inactivate the normal WDR45 allele is responsible for neurodegeneration seen in the rare female infants who manifest the X-linked pathologic variant (NBIA5), because their brains accumulate iron. If this disorder is caused by a truncating variant, only mosaic males, or females who are highly skewed favoring the normal allele, manifest the disease, as all other males and females with the variant die in utero. Affected females with hypomorphic variants often skew favoring the mutant allele (Table 1).8
DEGREE OF CELL SELECTION IS DETERMINED BY THE VARIANT
The X-linked form of Kabuki syndrome is caused either by point variants or deletions in KDM6A. Females with point variants often have Kabuki syndrome, whereas those with the larger deletions silence the X carrying the deletion; hence, they have less severe manifestations.31
Only a rare heterozygote with a variant in the PLP1 gene has symptoms of the myelin leukodystrophy associated with Pelizaeus–Merzbacher syndrome, and she invariably shows chance skewing favoring the mutant cells.32 In addition, during the central nervous system (CNS) development of heterozygotes, the oligodendrocytes with severe PLP1 disease alleles are negatively selected (apoptosis) in favor of wild type cells, resulting in skewed X-inactivation that is cell type specific.8
ESCAPE GENES INFLUENCE PHENOTYPE
Another factor that influences the clinical manifestations of X-linked variants in heterozygotes is the partial expression of genes from the inactive X chromosome.33 In addition to those genes in the pseudoautosomal region that are expressed on both sex chromosomes, more than 100 genes on the human X chromosome are expressed not only from the active X, but also to some extent from the silent X.34 They are referred to as escape genes. In fact, such genes do not really escape silencing, as they function to some extent, usually producing 10–30% of the level of transcripts made by the homologous allele on the active X. Yet, this little extra expression of a gene does influence phenotype. For example, male fetuses with pathologic variants causing orofacial digital syndrome all die in utero, unless they have a second X chromosome (i.e., Klinefelter syndrome); human females survive birth and die in their forties, usually from renal failure. However, female mice with the same variant outlive males by only several weeks as they die as neonates due to their polycystic kidneys. The important species difference is that humans, but not mice, partially express the OFD1 gene from their inactive X.13 This little extra gene activity is responsible for the difference in age of death of females of the two species. A little extra gene product from the inactive X can ameliorate the effects of an X-linked variant (see Table 2).
The extra product may not be available in every tissue, as there are tissue-specific differences in the expression of escape genes. In several females with a USP9X variant, pigment changes along the Blaschko lines and body asymmetry were observed, which is probably related to differential escape from X-inactivation between tissues35 and Table 1.
Yet, a little extra activity from the inactive X is not always helpful. It seems that expression of the Toll-like receptors on the inactive X is partly responsible for the impressive sex differences in some cases of autoimmune diseases like Lupus and Sjogren disease.36 Toll-like receptors, encoded by the X chromosome, are signaling pattern recognition receptors that are an integral part of innate immunity. The little extra activity may provide females with better protection against infectious agents, but it may make them more susceptible to autoimmunity. Such disorders are nine times as frequent in females than males and are also increased in Klinefelter XXY males, whereas as Turner females, even those taking estrogens, have the same risk as XY males. It is now thought that the excess manifestations of autoimmune disease in women are due to a complex of issues. Studies in mice show that increasing the expression of the X-linked Toll-like receptors in susceptible mice increases the expression of the autoimmune disorder.36 It has not yet been shown that affected females and Klinefelter males have greater expression of their Toll-like receptors than those women and XXY males who do not have the disorder.
Other relevant X-linked genes with potential to influence autoimmunity and that escape X-inactivation include CXCR3,37 KDM6A,38 and CXorf21, which has been shown to be more highly expressed in women and Klinefelter males than in normal men.39 In addition, the expression of XIST from the inactive X in lymphocytes differs significantly from that of other cells as the XIST RNA cloud is dispersed, leading to poorer Barr body formation.40 No doubt, it is the interaction of several factors that is responsible for the high incidence of autoimmunity associated with having two X chromosomes.
DIFFERENT DISORDERS FROM THE SAME GENE
What is increasingly apparent is that variants in a single gene can lead to differently named disorders, because of the specific effect of the variant on production of the gene product. Before variants were identified, diseases were classified by their phenotypes and named by the physician who reported the disease, based on its symptoms. Now, we know that many different phenotypic disorders may be due to variations in the same gene; their effect in the heterozygote reflects the severity of the variant, which has influenced the naming of the disorder. Table 1 shows some of these allelic disorders (in bold). Yet, in most cases, no matter the severity of the disease in males, the heterozygous female is better off than the hemizygous male.
Examples of allelic disorders are the different variants in the IKBKG gene, which can cause incontinentia pigmenti (IP) if dysfunction is severe, ectodermal dysplasia if dysfunction is moderate, and immunodeficiency 33 if it is mild. When the pathologic variant is severe (like IP), females may be the only ones that manifest the disorder, as males die in utero. On the other hand, when it is moderate, or mild (like immunodeficiency 33), women may completely escape its pathologic effects (Table 1).8
Another set of allelic disorders is caused by variants in the PRPS1 gene, responsible for syndromes of deafness (missense variant leading to reduced expression), gout (gain of function variant), and Charcot–Marie–Tooth disease (more severe reduction of expression), depending on the severity of the specific variant. Again, the heterozygous female always has a less severe phenotype than the hemizygous male. When the male is deaf, the female has high range hearing loss; when he has gout, she has no manifestations. However, when he has optic atrophy and neuropathies, she has much milder manifestations, or none at all (Table 1).8
There is a spectrum of allelic disorders caused by different variants in the filamin A gene; again females are always less affected than males with the same deleterious variants, and are the only ones affected with Melnick–Needles syndrome, because it is lethal in males (Table 1).8
There are also allelic disorders caused by different variations in the MECP2 gene. Rett syndrome results when the gene has a deletion or substitution variant that decreases its function; the loss of function causes a disorder that is usually lethal to unborn males, so that generally only females survive to manifest the disease. However, when the same gene is duplicated (Lubs X-linked intellectual disability [XLID], Table 1), the increased function permits males to survive and manifest the disorder, whereas the gene duplication provides the normal allele a selective advantage, so that females escape all manifestations, as the duplicated gene is always on the inactive X (Table 1).8
Another impressive example of the effect of allelic disorders on sex differences in expression of the disease are variants in the USP9X gene (Table 1).8,35 As point variants leading to truncation are lethal for the male fetus, the manifestations are confined to females. On the other hand, less severe nontruncated variants in the same gene cause hippocampal related mental retardation, hypotonia, and aggression in males, whereas carrier females have no abnormalities.
MANY X-LINKED GENES ARE ASSOCIATED WITH INTELLECTUAL DEFICIENCY
The proportion of X-linked variants causing intellectual deficits is striking. In addition to the many X-linked variants whose phenotypes include intellectual disability, there are many disorders that are specifically associated with X-linked intellectual deficiency, both syndromic and nonsyndromic; these are listed in OMIM as XLID followed by a number from 1 to 107. These disorders map from the telomere of the short arm to the last band on the long arm of the X. Although the role of the gene whose variant leads to the disorder is not always well defined, the genes responsible for X-linked intellectual disability are involved in many pathways. It seems that intelligence is the sum total of how all our genes are functioning. Malfunctions of genes in many pathways can interfere with intellectual capabilities. Extreme skewing of X-inactivation is frequently seen with XLID variants that permit male survival, and the variant is always found on the inactive X in females; consequently, they have normal intellectual function. Apparently, such disease-producing variants are toxic for expressing cells.
SUMMARY
Being mosaic for the function of their X-linked genes generally ameliorates the expression of X-linked deleterious variants in females. X-inactivation provides the opportunity to share gene products. If this is not possible, then cell selection may eliminate variant cells. Many of these variants affect intellectual ability. Females usually manifest their X-linked pathologic variants, if both alleles are disease variants, or if males with the same pathogenic variants are lost in utero. X-inactivation provides an enormous advantage to females with deleterious X-linked variants, most often enabling them to avoid disease manifestations, including intellectual disability, that affect males. Fortunately, chance skewing that favors mutant alleles is relatively rare (5%) in survivors. The female advantage is reflected by the 20% greater death rate for males at every stage postimplantation, until the age of >75 years, when more females die because there are fewer male survivors.41
The females susceptible to X-linked diseases are those who have more than one copy of a pathologic variant, or a relevant second variant, or a variant in an essential gene that does not permit males to survive (Table 2). Females are also susceptible to chance skewing favoring the mutant allele, or the effects of X chromosomal aberrations (i.e., translocations) and monozygotic twinning on inducing unfavorable skewing.42
The fact that more males are born than females (1.05:1) is also likely to be attributable to X-inactivation. In this case, preimplantation females are lost because of their greater dosage sensitivity in maintaining an active X.11 But if XX individuals successfully establish X-inactivation while in utero, then throughout their lifetime, they will benefit from the cellular mosaicism it produces.
References
Kessler E, Rivaud M, Vos M, van Veen T. Sex-specific influence on cardiac structural remodeling and therapy in cardiovascular disease. Biol Sex Differ. 2019;10:7.
Natri H, Garcia A, Buetow K, Trumble B, Wilson M. The pregnancy pickle: evolved immune compensation due to pregnancy underlies sex differences in human diseases. Trends Genet. 2019;35:478–488.
Nuzzi R, Scalabrin S, Becco A, Panzica G. Sex hormones and optic nerve disorders: a review. Front Neurosci. 2019;13:57.
Al-Salameh A, Chanson P, Bucher S, Ringa V, Becquemont L. Cardiovascular disease in type 2 diabetes: a review of sex-related differences in predisposition and prevention. Mayo Clin Proc. 2019;94:287–308.
Gogos A, et al. Sex differences in schizophrenia, bipolar disorder, and post-traumatic stress disorder: Are gonadal hormones the link?. Br J Pharmacol. 2019;176:4119–4135.
Migeon BR. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA. 2006;295:1428–1433.
National Center for Biotechnology Information. Gene. 2019. https://www.ncbi.nlm.nih.gov/gene.
OMIM. X-linked diseases. 2019. https://www.ncbi.nlm.nih.gov/omim/?term=X+linked+diseases.
Lyon MF. Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet. 1962;14:135–148.
Migeon BR. Choosing the active X: the human version of X inactivation. Trends Genet. 2017;33:899–909.
Migeon BR, Beer MA, Bjornsson HT. Embryonic loss of human females with partial trisomy 19 identifies region critical for the single active X. PLoS One. 2017;12:e0170403
Migeon BR. Females are mosaics: X inactivation and sex differences in disease. 2nd ed. New York: Oxford University Press; 2014.
Franco B, Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev. 2006;16:254–259.
Cox RP, Krauss MR, Balis ME, Dancis J. Evidence for transfer of enzyme product as the basis of metabolic cooperation between tissue culture fibroblasts of Lesch–Nyhan disease and normal cells. Proc Natl Acad Sci U S A. 1970;67:1573–1579.
Migeon BR, Sprenkle JA, Liebaers I, Scott JF, Neufeld EF. X-linked Hunter syndrome: the heterozygous phenotype in cell culture. Am J Hum Genet. 1977;29:448–454.
Migeon BR. Studies of skin fibroblasts from 10 families with HGPRT deficiency, with reference in X-chromosomal inactivation. Am J Hum Genet. 1971;23:199–210.
Migeon BR. X-linked hypoxanthine-guanine phosphoribosyl transferase deficiency: detection of heterozygotes by selective medium. Biochem Genet. 1970;4:377–383.
Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A. 1981;78:5066–5070.
Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am J Hum Genet. 2002;71:168–173.
Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ. et al. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet. 2013;22:1654–1662.
Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162:570–572.
Pinto LLC, Vieira TA, Giugliani R, Schwartz IVD. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14.
Beck M, Cox T. Why are females with Fabry disease affected? Mol Genet Metab Rep. 2019;21:100529.
Jacobs PA, Hunt PA, Mayer M, Bart RD. Duchenne muscular dystrophy (DMD) in a female with an X/autosome translocation: further evidence that the DMD locus is at Xp21. Am J Hum Genet. 1981;33:513–518.
Quan F, Janas J, Toth-Fejel S, Johnson DB, Wolford JK, Popovich BW. Uniparental disomy of the entire X chromosome in a female with Duchenne muscular dystrophy. Am J Hum Genet. 1997;60:160–165.
Burn J, Povey S, Boyd Y, Munro EA, West L, Harper K, et al. Duchenne muscular dystrophy in one of monozygotic twin girls. J Med Genet. 1986;23:494–500.
Trejo V, Derom C, Vlietinck R, Ollier W, Silman A, Ebers G, et al. X chromosome inactivation patterns correlate with fetal-placental anatomy in monozygotic twin pairs: implications for immune relatedness and concordance for autoimmunity. Mol Med. 1994;1:62–70.
Soltanzadeh P, Friez MJ, Dunn D, von Niederhausern A, Gurvich OL, Swoboda KJ, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord. 2010;20:499–504.
Plenge RM, Hendrich BD, Willard HF. A mutation in the XIST promoter in a family with nonrandom X chromosome inactivation. Am J Hum Genet. 1995;57S:A30.
Lau A, Brown C, Penaherrera MS, Langlois S, Kalousek D, Robinson W. Skewed X-chromosome inacctivation is common in fetuses or newborns associated with confined placental mosaicism. Am J Hum Genet. 1997;61:1353–1361.
Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC. et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124.
Scala M, Traverso M, Capra V, Vari MS, Severino M, Grossi S, et al. Pelizaeus–Merzbacher disease due to PLP1 frameshift mutation in a female with nonrandom skewed X-chromosome inactivation. Neuropediatrics. 2019;50:268–270.
Disteche CM. Escape from X inactivation in humans and mouse. Trends Genet. 1995;11:17–22.
Balaton BP, Dixon-McDougall T, Peeters SB, Brown CJ. The eXceptional nature of the X chromosome. Hum Mol Genet. 2018;27:R242–R9.
Reijnders MR, Zachariadis V, Latour B, Jolly L, Mancini GM, Pfundt R, et al. De novo loss-of-function mutations in USP9X cause a female-specific recognizable syndrome with developmental delay and congenital malformations. Am J Hum Genet. 2016;98:373–381.
Syrett C, Anguera M. When the balance is broken: X-linked gene dosage from two X chromosomes and female-biased autoimmunity. J Leukocyte Biol. 2019;106:919–932.
Oghumu S, Varikuti S, Stock J, Volpedo G, Saljoughian N, Terrazas C, et al. Cutting edge: CXCR3 escapes X chromosome inactivation in T cells during infection: potential implications for sex differences in immune responses. J Immunol. 2019;203:789–794.
Yuichiro I, Golden L, Noriko I, Matsukawa M, Ren E, Vincent T. et al. The X-linked histone demethylase Kdm6a in CD4+T lymphocytes modulates autoimmunity. J Clin Invest. 2019;130:3852–3863.
Harris VM, Koelsch KA, Kurien BT, Harley ITW, Wren JD, Harley JB, et al. Characterization of cxorf21 provides molecular insight into female-bias immune response in SLE pathogenesis. Front Immunol. 2019;10:2160.
Syrett C, Paneru B, Sandoval-Heglund D, Wang J, Banerjee S, Sindhava V. et al. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight. 2019;4:126751
Tarin JJ, Garcia-Perez MA, Hermenegildo C, Cano A. Changes in sex ratio from fertilization to birth in assisted-reproductive-treatment cycles. Reprod Biol Endocrinol. 2014;12:56.
Kristiansen M, Knudsen GP, Bathum L, Naumova AK, Sorensen TI, Brix TH, et al. Twin study of genetic and aging effects on X chromosome inactivation. Eur J Hum Genet. 2005;13:599–606.
Acknowledgements
I am grateful to readers Haig Kazazian, Hans Bjornsson, and Steven Salzberg in our McKusick-Nathans Department of Genetic Medicine for their helpful comments. Thanks also to Cate Kiefe for her adaptation of Fig. 1 from Current Opinion in Genetics & Development 2006;16:254–259, and to its authors, Brunella Franco and Andrea Ballabio, for permission to use it.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
B.R.M. declares no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Migeon, B.R. X-linked diseases: susceptible females. Genet Med 22, 1156–1174 (2020). https://doi.org/10.1038/s41436-020-0779-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0779-4
This article is cited by
-
Carrier frequency estimation of pathogenic variants of autosomal recessive and X-linked recessive mendelian disorders using exome sequencing data in 1,642 Thais
BMC Medical Genomics (2024)
-
Overcoming genetic and cellular complexity to study the pathophysiology of X-linked intellectual disabilities
Journal of Neurodevelopmental Disorders (2024)
-
Insights and implications of sexual dimorphism in osteoporosis
Bone Research (2024)
-
Trisomy silencing by XIST: translational prospects and challenges
Human Genetics (2024)
-
Characteristics and clinical evaluation of X chromosome translocations
Molecular Cytogenetics (2023)
