
Addendum to: “ACMG practice guideline: Genetic evaluation of short stature”. Laurie H. Seaver, MD and Mira Irons, MD; ACMG Professional Practice and Guidelines Committee Genetics in Medicine 11:465–470 (2009); https://doi.org/10.1097/GIM.0b013e3181a7e8f8; published online 02 April 2009.
This document was reaffirmed by the ACMG Board of Directors as of 27 October 2020 with the following addendum as a Focused Revision:
We conducted a comprehensive search of the literature published between 2009, when the previous guidelines were published, and May 2020. Keywords used in PubMed included “short stature,” “genetic evaluation,” “short stature microarray,” “short stature exome sequencing” using [All Fields] [TITLE-ABS-KEY] criteria. A total of 583 articles were found of which 539 primarily addressed the identification of genes associated with growth and gene defects associated with short stature. There were 44 articles regarding the genetic evaluation of short stature. We reviewed these articles and provide the following focused revision to the original document.
-
1.
We have updated the previously published algorithm for the genetic evaluation of short stature1 (Fig. 1) with the following alterations:
-
a.
Girls who show persistent or evolving short stature in childhood should have a karyotype included in their initial short stature/failure-to-thrive work-up as screening for Turner syndrome, which is often delayed until adolescence,2 resulting in missed opportunities for condition-specific interventions. A referral to endocrinology should be considered in early childhood, because a sex bias in short stature referrals has been found.3 In girls with persistent or evolving short stature, microarray would be indicated if Turner syndrome has been excluded.
-
b.
Chromosomal microarray (comparative genomic hybridization [CGH] and/or single-nucleotide polymorphism [SNP]) should be part of the initial genetic work-up for idiopathic short stature (ISS) and small for gestational age (SGA) with persistent short stature as well as syndromic short stature, since the yield of pathogenic and likely pathogenic copy-number variants (CNV) was reported as high as 10% in this population in one study.4 Multiple studies have reaffirmed use of microarray as first-line testing in patients with syndromic short stature with an average yield of 10–15%.5,6,7,8 It is important to note that SNP-based chromosomal microarray can document uniparental isodisomy, but not uniparental heterodisomy or methylation patterns.9,10 Therefore, further specific methylation or uniparental heterodisomy testing should be considered for any condition related to methylation defects (e.g., Silver–Russell syndrome, Temple syndrome).
-
c.
Rapid technological development has led to the discovery of an increasing number of novel genetic causes for short stature. Multiple genes that cause skeletal dysplasia have been implicated in cases of ISS and SGA with persistent short stature. Several genes associated with endocrinopathies, such as the growth hormone (GH)-insulin-like growth factor-1 (IGF-1) axis syndromes, have also been observed in children with ISS.11,12,13,14,15,16,17,18,19,20,21 Therefore, clinical phenotypes of short stature-associated syndromes are expected to expand, and molecular testing for children with short stature should be considered (particularly SHOX) even without overt signs of skeletal dysplasia or endocrinopathy.22,23
-
d.
Clinicians should explore the yield and other limitations of individual next-generation sequencing (NGS) panels and array technologies based on the data from the laboratory offering the testing. Clinicians should be aware of difficult-to-sequence regions, including genes located in highly homologous and repetitive regions.24 For example, the SHOX and GH1 genes are located in segmental duplication regions and NGS has a limited coverage and detection limitations.
-
e.
Further testing with clinical exome sequencing and referral to medical genetics should be considered for patients with the following features suggestive of a monogenic cause for short stature: significant short stature (height < -3 SD), facial dysmorphism, skeletal abnormalities, intellectual disability, microcephaly, multiple pituitary hormone deficiency, severe growth hormone deficiency, SGA with persistent short stature, family history of consanguinity, or family history of one parent with height < -2 SD.25,26,27,28 Current studies assessing diagnostic yield of exome sequencing for syndromic short stature with prior negative karyotype, microarray and NGS targeted panels is reported between 16.5% and 46%.29,30,31,32 Clinical genome sequencing has begun to be offered in select laboratories and can be considered if available. At this time, important considerations include the cost of this testing, insurance reimbursement, and lack of evidence that clinical genome sequencing has a significantly increased diagnostic yield compared with clinical exome sequencing.33 Additionally, clinical genome sequencing has not been studied specifically in any short stature cohort in the literature.
-
f.
Important resources for clinicians to utilize in the evaluation and management of patients with a genetic diagnosis that includes short stature include disease-specific growth charts (which can be found on CDC.gov or disease-specific organization websites), GeneReviews® and the ACMG and American Academy of Pediatrics (AAP) practice guidelines.
Fig. 1 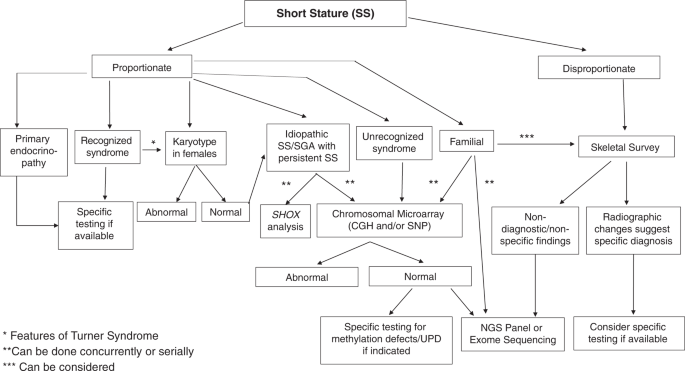
Algorithm for the genetic evaluation of short stature.
-
a.
-
2.
Because the ACMG 2009 short stature document1 does not meet the criteria for an evidence-based practice guideline by the ACMG (2014), it is now reclassified as a Clinical Practice Resource.

References
Seaver, L. H. & Irons, M. ACMG practice guideline: genetic evaluation of short stature. Genet. Med. 11, 465–470 (2009).
Grimberg, A. et al. Medically underserved girls receive less evaluation for short stature. Pediatrics. 127, 696–702 (2011).
Grimberg, A., Kutikov, J. K. & Cucchiara, A. J. Sex differences in patients referred for evaluation of poor growth. J. Pediatr. 146, 212–216 (2005).
Van Duyvendoore, H. A. et al. Copy number variants in patients with short stature. Eur. J. Hum. Genet. 22, 602–609 (2014).
Zahnleiter, D. et al. Rare copy number variants are a common cause of short stature. PLoS Genet. 9, e1003365 (2013).
Canton, A. P. M. et al. Genome-wide screening of copy number variants in children born small for gestational age reveals several candidate genes involved in growth pathways. Eur. J. Endocrinol. 171, 253–262 (2014).
Wit, J. M. et al. Copy number variants in short children born small for gestational age. Horm. Res. Paediatr. 82, 310–318 (2014).
Homma, T. K. et al. Recurrent copy number variants associated with syndromic short stature of unknown cause. Horm. Res. Paediatr. 89, 13–21 (2018).
Bumgarner, R. Overview of DNA microarrays: types, applications, and their future. Curr. Protoc. Mol. Biol. 22, Unit-22.1 (2013).
Del Gaudio, D. et al. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22, 1133–1141 (2020).
Vasques, G. A., Andrade, N. L. M. & Jorge, A. A. L. Genetic causes of isolated short stature. Arch. Endocrinol. Metab. 63, 70–78 (2019).
Wit, J. M., Kiess, W. & Mullis, P. Genetic evaluation of short stature. Best Pract. Res. Clin. Endocrinol. Metab. 25, 1–17 (2011).
Wang, S. R. et al. Large-scaled pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. J. Clin. Endocrinol. Metab. 98, E1428–E1437 (2013).
Romero, C. J., Mehta, L. & Rapaport, R. Genetic techniques in the evaluation of short stature. Endocrinol. Metab. Clin. North Am. 45, 345–358 (2016).
Wit, J. M. et al. Mechanisms in endocrinology: novel genetic causes of short stature. Eur. J. Endocrinol. 174, R145–R173 (2016).
Hattori, A. et al. Next generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocr. J. 64, 947–954 (2017).
Murray, P. G., Clayton, P. E. & Chernausek, S. D. A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol. 6, 564–574 (2018).
Finken, M. J. J. et al. Children born small for gestational age: differential diagnosis, molecular genetic evaluations, and clinical implications. Endocr. Rev. 39, 851–894 (2018).
Jee, Y. H., Baron, J. & Nilsson, O. New developments in the genetic diagnosis of short stature. Curr. Opin. Pediatr. 30, 541–547 (2018).
Yang, L. et al. Pathogenic gene screening in 91 Chinese patients with short stature of unknown etiology with a targeted next-generation sequencing panel. BMC Med. Genet. 19, 212 (2018).
Freire, B. L. et al. Multigene sequencing analysis of children born small for gestational age with isolated short stature. J. Clin. Endocrinol. Metab. 104, 2023–2030 (2019).
Marchini, A., Ogata, T. & Rappold, G. A. A track record on SHOX: from basic research to complex models and therapy. Endocr. Rev. 37, 417–448 (2016).
Funari, M. F. A. et al. Evaluation of SHOX defects in the era of next-generation sequencing. Clin. Genet. 96, 261–265 (2019).
Alkan, C., Sajjadan, S. & Eichler, E. E. Limitations of next-generation genome sequence assembly. Nat. Methods. 8, 61–65 (2011).
Waldman, L. A. & Chia, D. J. Towards identification of molecular mechanisms of short stature. Int. J. Pediatr. Endocrinol. 2013, 19 (2013).
Dauber, A., Rosenfeld, R. G. & Hirschhorn, J. N. Genetic evaluation of short stature. J. Clin. Endocrinol. Metab. 99, 3080–3092 (2014).
Guo, M. H. et al. Whole exome sequencing to identify genetic causes of short stature. Horm. Res. Paediatr. 82, 44–52 (2014).
Argente, J. Challenges in the management of short stature. Horm. Res. Paediatr. 85, 2–10 (2016).
Kim, Y. M. et al. High diagnostic yield of clinically unidentifiable syndromic growth disorders by targeted exome sequencing. Clin Genet. 92, 594–605 (2017).
Hauer, N. N. et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet. Med. 20, 630–638 (2018).
Huang, Z. et al. Genetic evaluation of 114 Chinese short stature children in the next generation era: a single center study. Cell. Physiol. Biochem. 49, 295–305 (2018).
Homma, T. K. et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J. Pediatr. 215, 192–198 (2019).
Thiffault, I. et al. Clinical genome sequencing in an unbiased pediatric cohort. Genet. Med. 21, 303–310 (2019).
Author information
Authors and Affiliations
Consortia
Ethics declarations
Disclosure
A.G. reports receiving a grant from Pfizer, Inc. for investigator-initiated research and is a consultant for the Pediatric Endocrine Society Growth Hormone Deficiency Knowledge Center, sponsored by Sandoz. The other authors have no disclosures or conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Correspondence: ACMG (documents@acmg.net)
Rights and permissions
About this article

Cite this article
Mintz, C.S., Seaver, L.H., Irons, M. et al. Focused Revision: ACMG practice resource: Genetic evaluation of short stature. Genet Med 23, 813–815 (2021). https://doi.org/10.1038/s41436-020-01046-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01046-0
