
Abstract
Purpose
Existing research suggests that while some laboratories report variants of uncertain significance, unsolicited findings (UF), and/or secondary findings (SF) when performing exome sequencing, others do not.
Methods
To investigate reporting differences, we created virtual patient–parent trio data by merging variants from patients into “normal” exomes. We invited laboratories worldwide to analyze the data along with patient phenotype information (developmental delay, dysmorphic features, and cardiac hypertrophy). Laboratories issued a diagnostic exome report and completed questionnaires to explain their rationale for reporting (or not reporting) each of the eight variants integrated.
Results
Of the 39 laboratories that completed the questionnaire, 30 reported the HDAC8 variant, which was a partial cause of the patient’s primary phenotype, and 26 reported the BICD2 variant, which explained another phenotypic component. Lack of reporting was often due to using a filter or a targeted gene panel that excluded the variant, or because they did not consider the variant to be responsible for the phenotype. There was considerable variation in reporting variants associated with the cardiac phenotype (MYBPC3 and PLN) and reporting UF/SF also varied widely.
Conclusion
This high degree of variability has significant impact on whether causative variants are identified, with important implications for patient care.
Similar content being viewed by others

INTRODUCTION
Widespread use of genomic sequencing in the clinical setting across a range of medical indications is leading to improvements in patient care.1,2,3 Yet relatively few studies4,5 have explored the analysis and reporting decisions made by laboratory scientists. These suggest that despite use of classification guidelines, there remains an element of subjectivity in variant analysis/interpretation.4,5 An interview-based study of 17 laboratory scientists across Europe, Australia, and Canada highlighted that variants might be reported differently depending not only on which scientist performs the analysis but also on their mood or how busy they are.4 While the study suggested decisions about which variants are reported are not usually left to one person, a survey of 21 US-based laboratories showed varying use of group discussion in case review between laboratories.4,6
There are considerable differences between laboratories in the types of variants they choose to report.4,6,7 Some, but not all, of the scientists interviewed report unsolicited findings when they are identified incidentally during the course of the analysis.7 Variation also exists between laboratories in the degree to which variants of uncertain significance (VUS) are reported, which appears to be based on both the analysis strategies used and also the laboratory’s overarching policies.4 The US-based survey also identified some divergence in laboratory reporting practices, particularly with the types of secondary findings they actively search for and report.6 In addition, a recent study interviewing 31 genetic health professionals suggested that reanalysis processes—where laboratories rerun previously analyzed data through their bioinformatic pipeline to check for new causative variants—are quite haphazard.8
Yet, to our knowledge, no research has been conducted to systematically analyze the actual reporting practices of laboratories worldwide. To address this, we invited laboratories to analyze “patient” sequence data and issue a report to the “referring clinician” (i.e., the research team).
MATERIALS AND METHODS
A virtual patient–parent trio was created by merging variants from patients into “normal” exomes. The virtual patient family was based on the genomic background of a genome-in-a-bottle (GIAB) trio (daughter NA12878, father NA12891, mother NA12891), which was enriched using the Agilent SureSelect v6 ES capture kit (elidS07604514), sequenced on the Illumina HiSeq 2500, and processed in a routine clinical setting. By substituting reads overlapping disease-causing variants from actual patients, the GIAB samples were used to create virtual patients. Eight variants were inserted into the existing genome sequence (Table 1). Two variants were relevant to the developmental delay and dysmorphic features present in the patient and her mother (HDAC8 and BICD2 genes). Two variants could have accounted for the cardiac symptoms (MYBPC3 and PLN genes). Four additional variants were included that were not related to the phenotype of the patient: a variant in the FLCN gene that can be considered an unsolicited finding (UF), a heterozygous variant in the recessive PAH gene, and two variants (one pathogenic variant and one VUS) in the MUTYH gene.
The GIAB BAM files were split into in- and off-target based on a BED file using samtools v1.29 with all variant positions that needed to be substituted in that specific sample: samtools view -b (GIAB_bam) -o (off-target.bam) -U (in-target.bam) -L (region.bed), with region.bed being all included disease variant positions. All reads (including mate pairs) within the off-target.bam were removed from the original FASTQ files, thereby creating stripped GIAB FASTQ files. For the patient BAM file, the opposite procedure was performed: samtools view -b (GIAB_bam) -o (in-target.bam) -U (off-target.bam) -L (region.bed). All reads (including mate pairs) within the in-target.bam were extracted from the original patient FASTQ files, thereby creating variant specific FASTQ files for each patient. The FASTQ files of each virtual patient was made by merging the stripped GIAB FASTQ and corresponding variant specific patient FASTQ files. The virtual patient FASTQ files were used as input for our in-house developed pipeline (IAP v2.7.0) providing the BAM and VCF files for these patients.10 FASTQ, BAM, and VCFs are available for download.11
A description of the virtual patient clinical history was developed, which included information about the parents and other family members developed to correspond with the variants merged into the existing exome data (Supplementary file 1). The patient’s phenotype included developmental delay, muscle weakness, dysmorphic features, and cardiac hypertrophy. A pedigree is also provided in the supplementary material (Supplementary file 2), although this was not sent to laboratories.
Recruitment
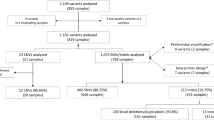
Potential participating laboratories were identified using three strategies: (1) searching Orphanet and the Genetic Testing Registry to identify laboratories performing exome sequencing, (2) existing connections made by K.L.V.G. through GeneMatcher, and (3) existing connections made by D.F.V. from another study.4 Potential participants were sent an introductory email inviting participation. Those interested were sent the clinical description of the patient and links to download the patient–parent trio data files and complete a questionnaire. They were also allocated a reference number to track their data across the questionnaires and patient report.
Data collection
Data collection involved two phases
Phase 1: Participating laboratories analyzed the data as per their standard procedures and issued a patient report, as they would to a referring clinician. Laboratories also completed an online laboratory characteristics questionnaire (Supplementary file 3), which asked the location and nature of their laboratory (i.e., commercial, hospital, university affiliated), number of exomes performed annually, quality assessment and pipeline information (accreditation, software usage for analysis, standard use of Sanger sequencing, etc.), whether they issue research or clinical reports, and their analysis and filtering strategies.
Phase 2: Once the report was received and the laboratory characteristics questionnaire was completed, participants were sent a reporting decisions questionnaire (Supplementary file 4), which investigated their rationale for reporting (or not reporting) each of the eight variants, how they classified each variant, and whether they actively searched for secondary findings. It also explored their policies for reporting variants in genes of uncertain significance (candidate genes), reanalysis of sequence data, and their criteria for actively searching for secondary findings.
Analysis
Fixed responses to both questionnaires were analyzed descriptively (i.e., how many laboratories reported each variant, etc.). A Chi-squared test of independence was performed to compare reporting between laboratories that included a clinical geneticist in the analysis and those that did not. Inductive content analysis was used to analyze free text questions to group responses into categories.12
Ethics statement
This study was performed in accordance with relevant guidelines and regulations and was approved by the SMEC Review Board (Social and Societal Ethics Committee), KU Leuven. Consent to use the genome-in-a-bottle resources can be found here: https://www.nist.gov/programs-projects/genome-bottle. Participants of invited laboratories gave written consent to participate in the study both by email and also by clicking “next” to begin each of the questionnaires.
RESULTS
Laboratory characteristics
Thirty-nine laboratories from 16 countries across 5 continents completed the questionnaires (Table 2). Laboratories most commonly described themselves as either hospital-affiliated (17), university-affiliated (7), or both (5). Five commercial laboratories participated. Most (33) laboratories performed a diagnostic exome analysis and 34 routinely performed in-house bioinformatic analyses. Forty-eight percent routinely outsourced their sequencing. Thirty laboratories indicated that their analysis involved a registered laboratory geneticist, laboratory director, or equivalent. Of these 30 laboratories, 18 stated their analysis also included a clinical geneticist, 12 included a technician, and 11 included an unregistered clinical scientist. Overall, 22 laboratories stated involvement of a clinical geneticist in the analysis. Research laboratories did not appear to report any differently to those performing diagnostic sequencing, so we have not separated the two.
Reporting practices and rationales
HDAC8
Of the 39 participating laboratories, 30 (77%) reported the variant in the HDAC8 gene, which was responsible for part of the patient’s primary phenotype (Cornelia de Lange syndrome) (Table 3). Of the nine that did not report this variant, most (7) indicated that this was because they used a filter which excluded the variant. The other two excluded the variant as it did not fit the pattern of inheritance.
BICD2
Twenty-six laboratories (67%) reported the variant in the BICD2 gene, which was responsible for the other component of the patient’s primary phenotype (spinal muscular atrophy with lower limb predominance) (Table 3). Of the 13 that did not report the variant in the BICD2 gene, almost half (6) stated they did not consider the variant to be the cause of the patient’s phenotype, either because they felt the phenotype did not match or they thought the phenotype was explained by the variant in the HDAC8 gene. Three laboratories stated they had identified the variant, but it is their policy not to report VUS. Two indicated they used a gene panel that did not include the variant, and two used a filter that excluded the variant. In total, 16 laboratories (46%) reported variants in both the HDAC8 and BICD2 genes.
MYBPC3
Thirty laboratories (77%) reported the variant in the MYBPC3 gene (Table 3). In many cases, the rationale for reporting was that they considered it to be relevant to the phenotype of the patient (12). Others reported this variant because they either actively search for genes in variants on the American College of Medical Genetics and Genomics (ACMG) list (12) or at least report UF in genes on the ACMG list if they are identified inadvertently (8). However, laboratories also commonly reported the variant because they report based on the age of onset of the potential phenotype (8), the possibility of prevention/surveillance (8), or the possibility of treatment (5). Five stated they report based on group consensus at team meetings. Of the nine (23%) that did not report the variant in the MYBPC3 gene, four used a filter that excluded the variant and two used a gene panel that did not include this gene.
PLN
Of the 16 laboratories (41%) that reported the variant in the PLN gene, many considered it to be relevant to the phenotype of the patient (9). Reporting based on the possibility of prevention/surveillance (4), the possibility of treatment (3), or based on group consensus at team meetings (3) were also prominent responses. Twenty-three laboratories (59%) did not report this variant, primarily because they had used a filter that excluded the variant (11).
FLCN
Fifteen laboratories (38.5%) reported the variant in the FLCN gene (Table 4). The most common rationale was due to the possibility of prevention/surveillance associated with reporting (9), with reporting based on the possibility of treatment (5), and reporting based on the age of onset of the potential phenotype (6) also listed as common reasons for reporting this variant. However, 24 (61.5%) did not report the variant in FLCN. This was often because their policy is not to report UF (13), although others either used a gene panel that did not include this gene (3) or used a filter that excluded the variant (4).
PAH
Only five laboratories (13%) reported the variant in the PAH gene. This was mainly because they routinely report heterozygous variant status in recessive disease when identified incidentally (2), or because they report heterozygous variant status based on severity of the recessive condition (2) or in genes related to the phenotype (2). Of the 34 (87%) that did not report this variant, this was primarily because their policy is not to report heterozygous variant status in recessive disease (22), or because they did not consider it to be the cause of the phenotype (9).
MUTYH
Fourteen (36%) reported at least one of the variants in the MUTYH gene. This was usually because they either actively search for genes in variants on the ACMG list (6) or report UF in genes on the ACMG list (7), with two specifying they only reported one of the variants because the second was a VUS. Of the 25 (64%) that did not report either of the variants, this was most commonly because they do not report VUS in genes unrelated to phenotype (14), although others used a filter which excluded the variant (6), or a gene panel that did not include this gene (2). Twenty-one laboratories (61.5%) actively searched for SF during the analysis.
Chi-squared tests of independence were performed to examine the relationship between reporting of each variant and the input of a clinical geneticist in the analysis. While the relationship was not significant for most variants (Supplementary file 5), the relationship was significant for the MYBPC3 variant, X2 (1, N = 38) = 7.2, P < 0.05. Laboratories with input of a clinical geneticist in the analysis were more likely to report the MYBPC3 variant.
Other reporting policies
Laboratories were asked to comment on three policies: (1) whether they typically report variants in candidate genes, (2) their criteria for actively searching for secondary findings, and (3) whether they reanalyze sequence data.
Candidate genes
Twenty-two (56%) laboratories said they report VUS in candidate genes (Table 5). Many suggested that they might report such a variant if there was evidence that it could match the phenotype. Some of the criteria that laboratories listed were if the variant was de novo, a homozygous loss-of-function pathogenic variant, when it is not found in control databases, when prediction tools suggest pathogenicity, when the residue is conserved, or where there are model organisms or pathway information.
Secondary findings
The most commonly stated criteria for proceeding with active searching was if the patient (or their legal guardian) has given consent (14) (Table 5). Yet seven indicated that they always search for SF, regardless of the request from the clinician or the age of the patient (7). Three only searched for SF in competent adult patients when requested by the clinician. Of those that do not actively search for SF, for a proportion this was based on either a national policy (2), a local/internal policy (3), or a lack of national consensus (2). Four (all from the same country) awaited results of a research study on SF before developing a policy.
Performing reanalysis
All 39 laboratories (100%) indicated that they perform reanalysis. However, the conditions under which they reanalyze and the degree of reanalysis that takes place varied (Table 5). Thirty-three will reanalyze patient data if requested by the referring clinician (84.6%), and 18 (46%) will issue a new report if a variant is reclassified (i.e., up- or downgraded from one class to another). Only 10 (26%) routinely reanalyze patient data, whereas 13 (33%) will reanalyze patient data if requested by the patient.
DISCUSSION
We identified considerable variation in reporting between laboratories across all eight variants, including those that were related to the patient’s phenotype.
Reporting related to the patient phenotype
The large difference in reporting on the variants in the HDAC8 and BICD2 genes we identified illustrates that the analysis decisions made by laboratory scientists influence what they do (or do not) report and that using a trio approach with strict filtering based on inheritance can negatively impact variant detection. In particular, it points to the tension faced by scientists who are forced to make tradeoffs. On the one hand, a more “open” analysis will identify many variants that require manual curation, which is more time-consuming for the laboratory scientist. While this method has a greater chance of including the causative variant—particularly when the phenotype is quite complex—it also has a greater chance of identifying unsolicited findings. On the other hand, performing a more targeted and phenotype-driven analysis may limit the number of variants identified, making curation more manageable and reproducible, yet may be too narrow and lead to potentially causative variants being missed. One potential strategy that could be employed is a stepwise approach where scientists could begin with a targeted phenotype-driven analysis and then move to a more open analysis if no causative variant is identified. One method that has been adopted by some laboratories requires the clinical geneticist to complete a requisition form where they must choose Human Phenotype Ontology (HPO) terms before the analysis begins. This enables the laboratory to develop a personalized gene panel analysis for each patient and ensures that phenotypic features the clinical geneticist thinks are relevant are not overlooked. Unfortunately, only one guideline addresses the need to update gene panels and internal phenotypic algorithms based on new and/or broadening phenotypes: the ACMG recommends evaluating gene panels for updates every 6 months.13 In addition, the PanelApp initiative—an open online platform for laboratories, clinicians and researchers—is attempting to address this to some extent.14 Importantly, information about the approach taken, such as whether a targeted gene list was used, should be provided on the report to inform referring clinicians about the potential limitations of the analysis.
Six laboratories stated that they did not consider the variant in the BICD2 gene to be the cause of the patient’s phenotype. Our study design did not allow for further elaboration of the rationale for this view. Yet, it would be interesting to know if this was because they had already identified the variant in the HDAC8 gene and therefore considered the case solved, even though it was only a partial explanation for the patient’s phenotype. This raises questions about how complete a report should be and to what degree laboratories are obligated to keep searching for potentially causative variants. Existing research suggests that patients with greater clinical complexity are more likely to have multiple potentially relevant genetic findings.15 Therefore, these types of cases could be flagged for additional investigation. Another critical question relates to genetic health professionals’ expectations for the extent to which laboratories continue to search for potentially causative variants to fully explain the phenotypic picture. While trained clinical geneticists are more likely to recognize that the unexplained component of the phenotype may have its own genetic cause, this is much less likely to be identified by other referring health professionals. As most laboratory scientists do not have such clinical expertise, input from a trained clinician prior to a report being issued may help combat this.
In addition, three laboratories indicated that they did not report the variant in the BICD2 gene because their policy is not to report VUS. This accords with the findings of previous studies that showed varying practices between laboratories with the reporting of VUS; some laboratories limit their reporting to pathogenic and likely pathogenic variants, whereas others report VUS in genes known to be related to the phenotype, or even candidate genes.4,16 Overall, position statements by professional bodies do not provide clear guidance on this point. The Canadian College of Medical Geneticists (CCMG) and European Society of Human Genetics (ESHG) do not address whether VUS should be reported17,18 and EuroGentest recommendations leave it to laboratories to decide.19 This is in contrast to a multidisciplinary Working Group consensus document that suggests that VUS that are identified in genes known to be related to the patient phenotype should be reported because it could be an answer to the clinical question.20 If this guidance was followed, the variant in the BICD2 gene should have been reported. Interestingly, of the 26 laboratories who reported this variant, 14 reported it as likely pathogenic and 3 as pathogenic. Based on the ACMG classification system criteria (Richards et al. 21), this variant can be classified as likely pathogenic (evidence PS2, PM2, PM5, and PP3). However, interviews with laboratory scientists across Europe, Australia, and Canada highlighted the subjective nature of variant interpretation that can result in different classifications of the same variant, even when standardized classification frameworks are used.4
The clinical patient description provided to laboratories also included an indication of a cardiac phenotype—mild cardiac hypertrophy on magnetic resonance imaging (MRI). Although in theory the cardiac phenotype could have been related to the HDAC8 gene variant, the family history was also suggestive of sudden cardiac death in both the patient’s paternal uncle and grandfather. Although the pathogenic variants in the PLN or MYBPC3 genes were reported by many laboratories, a large number of laboratories did not report them. This demonstrates that even though the laboratories all received the same clinical details, some determined that the cardiac phenotype was relevant to include in the analysis, whereas others did not. This reflects findings of another study in which variants in the GJB2 gene were not reported as likely to be the cause of hearing loss in the proband because it was not considered to be the primary phenotype.22 This difference has significant implications for patient care. Our finding that laboratories that had input of a clinical geneticist in their analysis were more likely to report the variant in the MYBPC3 gene suggests that using clinician-selected HPO terms to help guide the analysis may help laboratories determine which phenotypic features should guide the analysis and therefore what to report.
Variants unrelated to the patient phenotype
We included four variants that were unrelated to the patient’s phenotype to examine how laboratories dealt with UF. There was considerable variation in reporting the variant in the FLCN gene between laboratories, often based on laboratory policies not to report UF. This is another point on which there is a lack of consensus in guidelines and also varying practices between laboratories.7,16 Although the ESHG does not provide any specific guidance for the return of UF in children, they recommend that UF “indicative of serious health problems” identified in adults should be reported.18 The ACMG states that only variants that are actively searched for, such as in genes on their “minimum list” of 59 genes or additional genes for which consent is specifically obtained, should be reported in both adults and children.23,24 This list does not currently include FLCN and, as such, reporting of variants would not usually be supported. The American Society of Human Genetics (ASHG) and the CCMG suggest that when a UF identifies a serious condition that is actionable in childhood, and the condition is serious, this should be reported to parents, whether they agree to it or not.17,25 Birt–Hogg–Dubé syndrome carries with it a risk of renal cancer and early diagnosis has the potential for preventive measures to be enacted. However, as these symptoms often do not present until adulthood, the ASHG and CCMG recommendations do not apply.26 In contrast, the consensus document developed by the multidisciplinary working group mentioned previously suggests that UF for adult-onset diseases identified in minors should be returned in circumstances where it has “significant implications for the health of a family member, or the child, during their lifespan,” provided there is some treatment or prevention available.20 According to this recommendation, the UF in the FLCN gene should have been reported.
Most laboratories did not report the pathogenic variant in the PAH gene, most commonly because their policy is not to report heterozygous variant status in recessive disease. Professional guidelines often do not comment specifically on whether an unsolicited finding of a heterozygous variant status in recessive disease should or should not be reported. Rather, it is open to interpretation; while some may not perceive it to be actionable because it will not change disease management, others may consider it to have clinical utility because it can be used to inform future reproductive decisions. It is based on this rationale that a Working Group consensus document suggested that heterozygous variant status in recessive disease identified during diagnostic genomic sequencing should be reported, regardless of the age of the patient, provided informed consent is obtained prior to testing.20
Although most laboratories did not report variants in the MUTYH gene, 14 laboratories did report either one or both variants. Recommendations by the ACMG suggest that variants in genes on their list that follow an autosomal recessive inheritance (of which the MUTYH gene in our virtual patient is one) should only be reported if variants in both alleles are pathogenic or likely pathogenic. As such, variants in the MUTYH gene in our patient should not have been reported.
Of note, 21 laboratories actively searched for variants in the ACMG gene list. It is interesting that seven years after their initial recommendation, the ACMG is still the only professional body to endorse such testing. Yet, clearly laboratories in other countries are embracing these guidelines because, although four of the laboratories from the United States searched for SF, so too did laboratories from France, India, Israel, Estonia, China, Switzerland, Saudi Arabia, Russia, Brazil, and Australia.
This study has several limitations. Although we attempted to recruit laboratories using multiple strategies, and included respondents from 16 countries across 5 continents, our data set may not be representative of laboratories worldwide. We were not able to determine the response rate of our study because we cannot tell how many of the potential participants approached using GeneMatcher were actually laboratories (versus clinicians who were not eligible to participate). Finally, it is possible that the laboratories that chose to participate are more interested in improving their reporting practices, which may mean they are more cognizant of current classification and reporting guidelines than others.
Nevertheless, we have shown that reporting practices vary considerably across laboratories. Notably, a sizable number of laboratories did not identify variants that were responsible for the phenotype of the patient, which was often due to analysis strategy decisions made by laboratories and has important implications for patient care. Our findings also highlight that, despite considerable debate over a number of years, there remains a lack of uniformity on the reporting of unsolicited findings and whether secondary findings should be actively sought. This is unsurprising given the absence of consensus across professional bodies on these points. As such, further work to develop broader consensus for laboratories is warranted to ensure equity in patient care.
References
Stark Z, Lunke S, Brett GR, et al. Meeting the challenges of implementing rapid genomic testing in acute pediatric care. Genet Med. 2018;20:1554–1563.
Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–1887.
Vears DF, Sénécal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet. 2017;60:553–558.
Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076.
O’Daniel JM, McLaughlin HM, Amendola LM, et al. A survey of current practices for genomic sequencing test interpretation and reporting processes in US laboratories. Genet Med. 2017;15:575–582.
Vears DF, Sénécal K, Borry P. Reporting practices for unsolicited and secondary findings from next generation sequencing technologies: perspectives of laboratory personnel. Hum Mutat. 2017;38:905–911.
Vears DF, Senecal K, Borry P. Genetic health professionals’ experiences with initiating reanalysis of genomic sequence data. Fam Cancer. 2020;19:273–280.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993.
Ernst RF, Boymans S, van Roosmalen M, et al. UMCUGenetics/IAP: v2.5.0. 2017. https://doi.org/10.5281/zenodo.597225.
Elferink MG, Vears DF, Kriek M, et al. Analysis of laboratory reporting practices using a quality assessment of a 'virtual patient'. (data). 2020. https://doi.org/10.5281/zenodo.3754090.
Downe-Wamboldt B. Content analysis: method, applications, and issues. Health Care Women Int. 1992;13:313–321.
Bean LJH, Funke B, Carlston CM, et al. Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020;22:453–461.
Martin AR, Williams E, Foulger RE, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51:1560–1565.
Smith ED, Blanco K, Sajan SA, et al. A retrospective review of multiple findings in diagnostic exome sequencing: half are distinct and half are overlapping diagnoses. Genet Med. 2019;21:2199–2207.
Hehir-Kwa J, Claustres M, Hastings RJ, et al. Towards a European consensus for reporting incidental findings during clinical NGS testing. Eur J Hum Genet. 2015;23:1601–1606.
Boycott K, Hartley T, Adam S, et al. The clinical application of genome-wide sequencing for monogenic diseases in Canada: position statement of the Canadian College of Medical Geneticists. J Med Genet. 2015;52:431–437.
van El CG, Cornel MC, Borry P, et al. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013;21:S1–S5.
Matthijs G, Souche E, Alders M, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24:1515.
Vears DF, Sénécal K, Clarke AJ, et al. Points to consider for laboratories reporting results from diagnostic genomic sequencing. Eur J Hum Genet: EJHG. 2018;26:36–43.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015:17;405–424.
Brownstein CA, Beggs AH, Homer N, et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014;15:R53.
Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255.
Green RC, Berg JS, Grody M,WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574.
Botkin JR, Belmont JW, Berg JS, et al. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 2015;97:6–21.
Ather H, Zahid N. Recurrent renal cancer in Birt–Hogg–Dube syndrome: a case report. Int J Surg Case Rep. 2018;42:75–78.
Acknowledgements
D.F.V. is a Postdoctoral Research Fellow of the Research Foundation–Flanders (FWO Vlaanderen) and also acknowledges the infrastructure funding received from the Victorian State Government through the Operational Infrastructure Support (OIS) Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Vears, D.F., Elferink, M., Kriek, M. et al. Analysis of laboratory reporting practices using a quality assessment of a virtual patient. Genet Med 23, 562–570 (2021). https://doi.org/10.1038/s41436-020-01015-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01015-7
Keywords
This article is cited by
-
Ethics of artificial intelligence in prenatal and pediatric genomic medicine
Journal of Community Genetics (2023)
-
Opportunistic genomic screening. Recommendations of the European Society of Human Genetics
European Journal of Human Genetics (2021)
