
Abstract
Purpose
Variants in the DNA mismatch repair (MMR) gene MSH6, identified in individuals suspected of Lynch syndrome, are difficult to classify owing to the low cancer penetrance of defects in that gene. This not only obfuscates personalized health care but also the development of a rapid and reliable classification procedure that does not require clinical data.
Methods
The complete in vitro MMR activity (CIMRA) assay was calibrated against clinically classified MSH6 variants and, employing Bayes’ rule, integrated with computational predictions of pathogenicity. To enable the validation of this two-component classification procedure we have employed a genetic screen to generate a large set of inactivating Msh6 variants, as proxies for pathogenic variants.
Results
The genetic screen-derived variants established that the two-component classification procedure displays high sensitivities and specificities. Moreover, these inactivating variants enabled the direct reclassification of human variants of uncertain significance (VUS) as (likely) pathogenic.
Conclusion
The two-component classification procedure and the genetic screens provide complementary approaches to rapidly and cost-effectively classify the large majority of human MSH6 variants. The approach followed here provides a template for the classification of variants in other disease-predisposing genes, facilitating the translation of personalized genomics into personalized health care.
Similar content being viewed by others
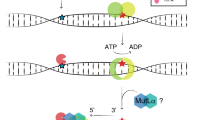

INTRODUCTION
Lynch syndrome (LS, OMIM 120435) is an autosomal dominant cancer predisposition, caused by a germline defect in a single allele of one of the mismatch repair (MMR) genes MSH2, MSH6, MLH1, or PMS2.1,2,3 Sporadic somatic loss of the wild-type allele results in cellular MMR deficiency. The mutator phenotype that results from the inability to correct DNA replication errors lies at the origin of LS-associated colorectal and endometrial cancers.1,2
Missense variants comprise a significant fraction of the genetic variants identified in the MMR genes, particularly in MSH6.3 In case such a variant cannot be classified as (likely) pathogenic or (likely) non-pathogenic, personalized health care for affected families cannot be implemented.2,4
The Variant Interpretation Committee (VIC) of the International Society for Gastrointestinal Hereditary Tumors (InSiGHT; https://www.insight-group.org) has devised the use of qualitative or quantitative integration of evidence to classify variants in the MMR genes, employing standards set by the International Agency for Research on Cancer (IARC).5 Unfortunately, classification of MSH6 variants has proven particularly difficult owing to the relatively low penetrance of cancer in LS patients with a proven MSH6 defect, which complicates the use of cosegregation as a diagnostic tool.5,6,7 For this reason, the VIC has been able to classify only a few MSH6 variants as pathogenic (IARC class 5, probability of pathogenicity >0.99), likely pathogenic (IARC class 4, probability of pathogenicity > 0.95), likely not pathogenic (IARC class 2, probability of pathogenicity < 0.05), or not pathogenic (IARC class 1, probability of pathogenicity < 0.001), with associated clinical recommendations.4,6 Therefore, the large majority of variants in MSH6 remain variants of uncertain significance (VUS; IARC class 3, probability of pathogenicity between 0.05 and 0.95).4,5,6
Functional assays may strongly contribute to improved classification of MMR gene variants.4,8,9,10,11 We have developed the complete in vitro MMR activity (CIMRA) assay to quantify the functional activity of variants in MMR genes.12,13,14,15 The assay can be performed in a few days using common laboratory equipment and only requires information on the variant (Fig. 1a). Such a functional assay–based classification procedure can only be transferred to the clinic following thorough calibration and validation. Calibration involves the regression of the assay output against the clinical odds in favor of pathogenicity (odds path) of a set of variants that have previously been securely classified by using clinical criteria only. The resulting regression formula converts the CIMRA assay result into odds path for the CIMRA assay, the variable that can be combined, using Bayes’ rule, with other calculated probabilities of pathogenicity, such as computational analysis, into a posterior probability (Posterior-P) of pathogenicity. The subsequent determination of the sensitivities and specificities of such a two-component classification procedure requires an unrelated validation set comprised of independently classified variants. We have recently followed a similar approach to develop a procedure to classify variants in MSH2 and MLH1.15 Unfortunately, because insufficient classified MSH6 variants are available, validation of a functional assay–based predictive procedure for variants in MSH6 has been extremely challenging.

(a) Outline of the CIMRA assay. (b) Relative repair efficiencies for MSH6 missense variants from the InSiGHT database, classified based on clinical criteria alone. Variants are ranked according to their mean CIMRA assay activity. The p.G1139S variant is included in every experiment as a (technical) repair-deficient control. Variants are colored according to their International Agency for Research on Cancer (IARC) classification (see figure for legend). Bars represent mean ± S.E.M. of >3 experiments. (c) Regressions of the CIMRA assay training values against odds in favor of pathogenicity. The y-axes of these graphs display probability of pathogenicity rather than Log(odds in favor of pathogenicity) to emphasize sigmoid calibration bounded at probabilities of 1.00 and 0.00. (d) Relative repair efficiencies for InSiGHT/ClinVar database-derived (likely) benign MSH6 missense variants (blue bars) as determined in the CIMRA assay. Variants are ranked according to their mean CIMRA assay activity. Bars represent mean ± S.E.M. of >3 experiments. MMR mismatch repair, PCR polymerase chain reaction, WT wild type.
Here, we have used the available clinically classified MSH6 variants to calibrate the CIMRA assay output and allow its Bayesian integration with previously calibrated and validated computational analysis into a two-component classification procedure. Then, we addressed the shortage of classified variants for validation purposes by generating a large number of in vivo inactivating Msh6 variants in a cell-based genetic screen. We have extensively characterized these variants, using cellular and biochemical analyses, to confirm their suitability as a proxy for pathogenic human variants. This has enabled the validation of the two-component classification procedure. Moreover, our finding that many inactivating variants identified in the genetic screen match human MSH6 VUS listed in variant databases supports their classification as pathogenic.
MATERIALS AND METHODS
Selection of classified missense substitutions for CIMRA assay calibration
In July 2017 we reviewed the InSiGHT variant database (http://insight-group.org/variants/database) for MSH6 variants that, by using clinical criteria alone, were classified as IARC class 4/5 or as class 1/2.5 We excluded those variants that had been used for calibration of the computational prior probability of pathogenicity (Prior-P).16 This resulted in a set of 24 variants. Since this number appeared insufficient for a robust calibration, we added 7 variants that have been classified as class 3 (VUS), although with observational data ≥3-fold evidence in favor of pathogenicity or ≥3-fold evidence against pathogenicity (Table S1).
Complete CIMRA assays
CIMRA assays of MSH6 variants were carried out as described,15,17 with a change of the use of nuclear extracts. To enable the production of highly active nuclear extracts,18 we generated MSH2 and MSH6 double-deficient HeLa cells. Briefly, cells were made MSH2-deficient with a CRISPR/Cas9 construct; these cells were selected using 6-thioguanine (20 µM). In these cells we also disrupted MSH6, using CRISPR/Cas9; MSH6-deficient clones were identified by polymerase chain reaction (PCR). Loss of both genes was verified by PCR and western blotting using Msh6 antibody ab14204 (Abcam).19 Detailed protocols and primer sequences are available upon request.
Regression for CIMRA assay calibration
For CIMRA assay calibration, InSiGHT observational odds in favor of pathogenicity in the form of Log10(Odds Path) was treated as the dependent variable.15 The normalized CIMRA assay values of the same variants were treated as the independent variable. We then performed linear regression on Log10(Odds Path) versus CIMRA assay values, whereby the use of Log(Odds) as the dependent variable constrains the resulting regression equations to produce probabilities between 0.00 and 1.00.
Computational analyses and Bayesian integration
Computational analyses predicting the probability of pathogenicity for each variant were performed using the calibrated and validated programs MAPP and PolyPhen-2, as previously reported.16 We used the resulting values as the Prior-P, setting upper and lower caps for Prior-P values at 0.10 and 0.90, to avoid classification as class 1/2 or class 4/5, based on computational prediction alone.16 The Prior-P is amenable to quantitative integration with the calibrated CIMRA assay results, to obtain a Posterior-P.5,20 Such a two-component classification procedure has previously been performed to integrate computational results with clinical parameters (such as segregation and tumor pathology), based on odds path.21,22
Generation of a set of independently classified variants for validation
To compile an independent validation set we reviewed the content of both the InSiGHT and ClinVar variant databases (https://www.ncbi.nlm.nih.gov/clinvar) for MSH6 variants that had met ClinVar or InSiGHT classification as (likely) benign/not pathogenic or (likely) pathogenic. We excluded variants that were used for calibration of the CIMRA assay or of the Prior-P,16 resulting in 18 remaining (likely) benign variants (Table S3).
No new, independently classified, class 4/5 variants were obtained from the databases. To obtain such variants we performed a genetic screen, essentially as described for Msh2.19 Briefly, we used the mutagen N-ethyl-N-nitrosourea (ENU; Sigma-Aldrich, St. Louis, MO) to introduce random substitution variants in Msh6-heterozygous mouse embryonic stem (mES) cells.23 Prior to use these cells were authenticated by PCR and mycoplasm testing. Since these cells are diploid for all other MMR genes than Msh6, MMR-deficient clones that were selected using 6-thioguanine (6-TG, Sigma-Aldrich) were expected to have lost the single Msh6 allele, rather than both copies of one of the other three MMR genes.24 Surviving clones were screened against inadvertent loss of heterozygosity of the Msh6 wild-type allele, rather than an ENU-induced substitution variant, by allele-specific PCR. We then screened against clones that did not express full-length Msh6 complementary DNA (cDNA), e.g., with nonsense or splice variants, by western blotting.23 To identify the ENU-induced substitutions that had inactivated the single wild-type Msh6 allele by a single missense variant, we sequenced Msh6 cDNA from the remaining clones. All primer sequences and PCR protocols are available upon request.
Assays to provide mechanistic insights in inactivating Msh6 substitutions obtained by a genetic screen
Microsatellite instability (MSI) was analyzed after PCR amplification of mononucleotide microsatellite mBAT-37 on ~50 subclones of variants each cell line tested.25 Fragment lengths were analyzed using GeneMarker software (Softgenetics).
Methylation tolerance was determined as follows: cells were treated for 1 hour with N-methyl-N’-nitro-N-nitrosoguanidine (MNNG; Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO), in increasing concentrations. O6-benzylguanine (40 µM; Sigma-Aldrich), an inhibitor of the repair protein methyltransferase, was added during treatment. After 3 days, IC50s were derived after counting of surviving cells.
Electrophoretic mobility shift assays were carried out using extracts from the variant cell lines, essentially as described.19,26 Oligonucleotides 5′-AGCTGCCAAGCACCAGTGTCAGCGTCCTAT-3′ and 5′-AGCTGCCAGGCACCAGTGTCAGCG TCCTAT-3′ were labeled at the 5′ end using γ-32P adenosine triphosphate (ATP) and polynucleotide kinase, and were both annealed to oligonucleotide 5′-ATAGGACGCTGACACTGGTGCTTGGCAGCT-3′ to generate a matched and a G·T mismatched (underlined) probe, respectively. Then, 170 fmol double-stranded oligonucleotide was incubated with 20 μg extract in DNA binding buffer (12% [vol/vol] glycerol, 20 mM Hepes/KOH pH 7.9, 100 mM NaCl, 1 mM DTT, and 0.1 mM EDTA, 0.05 μg/μL Poly[dIdC]) and 425 fmol unlabeled, matched oligonucleotide) for 20 minutes at 37°C in a total volume of 20 μL. For challenge experiments, ATP (final concentration 0.5 mM) was added 10 minutes after addition of the DNA probe. The reaction mixture was subjected to electrophoresis in a 4% polyacrylamide:bisacrylamide (29:1) gel in 0.5× TBE buffer containing 5% glycerol. Gels were dried, signals were visualized using a Cyclone Plus phosphor imager (PerkinElmer), and images were analyzed using OptiQuant software. All mismatch binding assay results were confirmed on independent cell extracts.
Sensitivity and specificity at the classification thresholds
Probabilities of pathogenicity were calculated following Bayesian integration of the computational Prior-P with the CIMRA assay values. Variants analyzed as having a Posterior-P of pathogenicity >0.95 were classified as class 4/5 whereas variants with a Posterior-P of pathogenicity <0.05 were classified as class 1/2. All other variants were classified as class 3 (VUS).
Sensitivities and specificities of the two-component (computational analysis and CIMRA assay) classification procedure were investigated using an independent validation set of variants that consisted of inactivating variants, generated in the genetic screen, and of benign variants derived from the ClinVar and InSiGHT databases. Sensitivity of the two-component procedure for class 4/5 variants was estimated as (# true positives)/(# inactivating variants). Specificity was estimated as (# true negatives)/(# [likely] benign variants).
Sensitivity of the two-component procedure for class 1/2 variants, derived from the ClinVar and InSiGHT databases, was estimated as (# true positives)/(# [likely] benign variants). Specificity was estimated as (# true negatives)/(# inactivating variants).
Multilaboratory assessment of CIMRA assay reproducibility
Variants used for in the multilaboratory assessment of CIMRA assay reproducibility were selected from the variants included in the calibration effort based on differential activities in the assay and differential positions within MSH6.
CIMRA assays were performed according to a protocol provided by Leiden University Medical Center (LUMC). Participating labs received technical support by email, when needed. Reagents (e.g., buffer and nuclear extract-containing CIMRA assay mix, substrate plasmid, etc.) were prepared at the LUMC and distributed to participating labs by mail. All commercially available components (e.g., TNT Quick Coupled Translation kit, Pfx Platinum polymerase, etc.) were purchased by the participating laboratories.
RESULTS
CIMRA assay calibration for quantitative data integration
To enable the integration of CIMRA assay results with other quantitative data, such as sequence analysis–based computational algorithms,5,20 we determined MMR activity of a calibration set consisting of 24 MSH6 missense variants selected from the InSiGHT database that had previously been securely classified using clinical criteria only (Fig. 1b and Table S1). CIMRA assay results were largely concordant with their previously assigned class (Fig. 1b and Table S1). Notably, all pathogenic variants displayed an activity in the CIMRA assay that was <25% of the wild-type control. To increase the power of the subsequent regression we included 7 VUS that had observational data providing ≥3-fold evidence in favor of, or against, pathogenicity (Fig. 1b and Table S1). We then performed linear regression of the CIMRA assay results of these 31 variants against log-transformed clinical odds in favor of pathogenicity, to derive a regression equation [Log10(Odds Path) = −0.0303508 (% activity) + 1.845465] (Fig. 1c). This equation enables conversion of CIMRA assay activities into odds in favor of pathogenicity—the variable that can be combined with other quantitative variables, using Bayes’ rule, to calculate a Posterior-P.4,5,6,15,16,20,27
We argued that, while loss of functional activity presumably predicts pathogenicity of a variant, the in vitro nature of the CIMRA assay might lead to false-negative results, e.g., when the variant would destabilize the protein in vivo. We therefore used Bayes’ rule to quantitatively integrate the CIMRA assay–based odds with a Prior-P of pathogenicity derived from sequence alignment-based algorithms that have been previously calibrated.16 This yielded a Posterior-P of pathogenicity, required for classification. All 11 variants from the calibration set that had been clinically classified as class 4/5 by InSiGHT were classified as pathogenic (class 5) by the two-component classification. Conversely, of 13 variants previously classified as class 1/2, the two-component classification classified 9 as (likely) not pathogenic (class 1/2), while classifying 4 as VUS (class 3). Importantly, no variants were misclassified (Table S1). The seven VUS were included for calibration purposes only and therefore have not been further classified.
Following recent guidelines established by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP), functional assays are considered to provide “strong” evidence in favor of or against pathogenicity (PS3/BS3).4 After CIMRA assay calibration and quantitative modeling of the ACMG/AMP criteria, the calculated CIMRA values can fall into any of six strength of evidence categories in favor of or against pathogenicity, as defined by ACMG/AMP (Table S2).4,27
A genetic screen for the identification of inactivating Msh6 missense variants
Validation of the two-component procedure should be performed on an independent set of variants, securely classified in the absence of functional or computational data.28 We were able to extract 18 additional MSH6 class 1/2 variants from the InSiGHT and ClinVar databases that were not present in the calibration set. In agreement with their classification, 17/18 variants showed activities greater than 60% of wild type in the CIMRA assay (Fig. 1d). Bayesian integration of the calibrated CIMRA assay results with the Prior-P corroborated the clinical classification of 14 variants as class 1/2, while four variants could not be classified using the two-component approach (Table S3). Thus, the two-component classification has a high sensitivity for class 1/2 variants.
Unfortunately, all MSH6 variants that, based on clinical data, were classified as class 4/5 in InSiGHT/ClinVar had already been employed in the calibration set and could therefore not be used for validation of the two-component classification (Fig. 1b). To still obtain in vivo inactivating MSH6 variants for this aim we employed a genetic screen in mouse embryonic stem cells (Fig. 2a), essentially as previously described for Msh2.19 This procedure resulted in the retrieval of 43 variant cell lines, each containing a random inactivating amino acid substitution in Msh6. In these 43 cell lines, 38 residues were affected; at 5 of these residues two different substitutions were identified, in different clones (Fig. 2b and Table S4). Of the 43 substitutions, 41 involved an amino acid that is conserved between mice and humans (Fig. S1 and Table S4).

(a) Pipeline to generate mouse embryonic stem (mES) cell lines that carry inactivating Msh6 missense variants. I. A mES cell line, heterozygous for Msh6 (Msh6+/−), is subjected to mutagenic treatment with ENU. II. Cells that have acquired 6-TG tolerance, by loss of heterozygosity at Msh6 (Msh6+/−), owing to an ENU-induced inactivating variant at a critical residue in the monoallelic Msh6 gene (Msh6M/−), or by an inactivating variant at the monoallelic Hprt gene (Hprt-) are selected using two brief 6-TG selections. III. The (unwanted) Hprt-deficient clones are eliminated by culture in HAT-supplemented medium. IV. Inadvertent clones that have lost the wild-type Msh6 allele by loss of heterozygosity (LOH) (rather than by a missense substitution) are excluded by using an allele-specific polymerase chain reaction (PCR). V. The inactivating substitution in the remaining clones is identified by sequence analysis. A “reverse diagnosis catalog” is compiled that lists inactivating substitutions at Msh6 as a proxy for pathogenic human variants (Fig. 4). (b) Representation of all inactivating missense substitutions in Msh6, identified in this screen. The top and bottom panels show alignments of the mismatch binding and ATPase domains of human and mouse Msh6, highlighting residues that were mutated in the genetic screen (orange boxes). Phe-X-Glu: mismatch-contacting loop. The middle panel displays all other inactivating Msh6 substitutions. Numbers reflect amino acid numbering of mouse Msh6. (c) Microsatellite instability (MSI) analysis. The size of each sphere is proportional to the relative number of subclones with the indicated mBAT-37 PCR fragment length. All mutants from the validation panel display MSI (p < 0.05 compared with the Msh6+/− line). (d) Tolerance of Msh6 mutant mES cell lines to the methylating drug N-methyl-N′-nitro-N-nitrosoguanidine. Bars represent mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001 (one-tailed Student’s t test) compared with the parental Msh6+/− line.
Genetic screens enable the identification of pathogenic human MSH6 variants
We argued that our genetic screen, in addition to allowing the validation of our two-component classification procedure, might yield inactivating Msh6 variants that coincide with previously identified, human MSH6 variants. This would directly support the pathogenicity of such variants. Indeed, 13 inactivating variants identified in our genetic screen were also found in the InSiGHT and ClinVar databases (Table S4). These included two alleles that had been classified by the InSiGHT VIC as class 4/5 and that were also used by us for the calibration of the CIMRA assay (p.L449P, p.G686D; Fig. 1b), as well as an allele we use as a “dead” control for the assay (p.G1139S; Fig. 1b, d).12,17 Importantly, none of the 41 inactivating alleles identified in the genetic screen had been classified by InSiGHT or ClinVar as class 1/2.
We wanted to further substantiate loss of in vivo MMR activity specifically for the eight genetic screen-derived cell lines of which the inactivating Msh6 variants matched variants that are listed in the InSiGHT database (Table S4). To this aim we first investigated microsatellite instability (MSI), a hallmark of MMR-deficient cancers.29 Indeed, all eight variant cell lines displayed MSI (Fig. 2c). In addition to a spontaneous mutator phenotype, MMR deficiency causes tolerance of methylating agents.30 As expected, all eight lines displayed strong tolerance of the methylating drug MNNG (Fig. 2d).
Inactivation of Msh6 function can be caused by different molecular defects
To pinpoint the molecular defects in Msh6 protein function of the variants identified in the genetic screen, and to further validate these as a proxy for pathogenic MSH6 variants, we performed biochemical analyses. MMR is initiated by binding of the MSH2/MSH6 heterodimer to a mismatched nucleotide pair.31,32,33 This induces ATP binding by both proteins, provoking a conformational change that converts the heterodimer into a clamp that slides on the DNA and binds MLH1/PMS2.34 We argued that the 43 inactivating variants from the genetic screen might either display a defect in any of these activities of Msh2/Msh6, or destabilize the Msh2/Msh6 heterodimer.
Western blotting of all inactivating Msh6-variant cell lines revealed that levels of the inactive Msh2/Msh6 proteins varied between the 43 different cell lines, supporting destabilization of some mutant proteins (Fig. 3a and Table S4). We then tested the ability of all mutated Msh2/Msh6 proteins to bind to mismatched oligonucleotides, employing electrophoretic mobility shift assays with extracts from the corresponding variant cell lines. The majority of the Msh6 variants displayed either partial or complete loss of mismatch binding, in many cases coinciding with reduced protein levels (Fig. 3a, b, Table S4).

(a) Western blot analysis of total lysates from (Msh6-variant) ES cells. Pcna serves as a loading control. In addition to Msh6 we also probed for Msh2. Since Msh2 stability depends on Msh6, Msh2 proteins levels are a surrogate marker for Msh6 protein stability. (b) Binding of control and Msh6-variant proteins to a G·T mismatch within a double-stranded oligonucleotide probe in an electrophoretic mobility shift assay. Bars represent mean ± S.E.M. of >3 experiments. (c) Adenosine triphosphate (ATP)-induced mismatch release of Msh6-variant proteins in an electrophoretic mobility shift assay. ATP (0.5 mM) was added after allowing proteins to bind to the probe. Bars represent mean ± S.E.M. For the purpose of clarity, all (-) ATP reactions are normalized to 1 and all (+) ATP reactions are normalized to their respective (-) ATP reactions. Bars represent mean ± S.E.M. of >3 experiments. (d) Msh6-mutant proteins deficient for ATP-induced release (Fig. 3c) were challenged with higher amounts of ATP in an electrophoretic mobility shift assay. ATP, in various concentrations, was added after allowing proteins to bind to the probe. Bars represent mean ± S.E.M. For the purpose of clarity, all (-) ATP reactions are normalized to 1 and all (+) ATP reactions are normalized to their respective (-) ATP reactions. Bars represent mean ± S.E.M. of >3 experiments. (e) Relative repair efficiencies, as determined in the complete in vitro MMR activity (CIMRA) assay, for human MSH6 missense variants, corresponding to inactivating murine variants identified in the genetic screen. Variants are ranked according to their mean CIMRA assay activity. Bars represent mean ± S.E.M. of >3 experiments/variant. The human, not the mouse, numbering of the variants is shown. The numbers at the bottom of the figure indicate the International Agency for Research on Cancer (IARC) class for every variant resulting from our calibrated two-component classification (Table S5).
Finally, we tested ATP-induced sliding clamp formation of those Msh2/Msh6 variants that had retained significant levels of mismatch binding (Fig. 3b and Table S4). In contrast to Msh2/Msh6-proficient cells, the addition of 0.5 mM ATP failed to induce release in extracts from cells expressing Msh6 variants p.N1134K, p.M1135K, p.G1137D, p.G1137S, and p.T1217I, explaining their defect in MMR (Fig. 3c, d, Table S4). Thus, these variants display a specific defect in sliding clamp formation.
In conclusion, using these specific biochemical assays, we have obtained insights into the molecular cause of MMR deficiency for 38 of the 43 variant alleles but were unable to do so for 5 substitutions (p.Q484K, p.Q484R, p.D1211E, p.D1211G, and p.H1246R). The observation that two of these amino acids each show two independent substitutions in different MMR-deficient cell lines suggests that both amino acids nevertheless are crucial for Msh6 function. We therefore infer that these variants may be defective in other characteristics of Msh2/Msh6.
The thorough biochemical characterization of the 43 inactivating variants produced in the genetic screen has (1) confirmed their causality for MMR deficiency, (2) enabled to pinpoint different biochemical defects as the cause of MMR deficiency of these (and, by inference, also LS-associated) variants, (3) demonstrated that the screen has provided an independent tool to directly assign pathogenicity to human MSH6 VUS that carry the identical substitution, and (4) demonstrated that the genetic screen-derived pathogenic variants serve as bona fide proxies for pathogenic human variants, warranting their suitability for validation of the two-component classification.
Validation of the two-component classification procedure
To validate the two-component classification, we used human analogs of (mouse) inactivating variants, identified in the genetic screen. From these variants we omitted Msh6 p.L872P and p.L1154R, as the mutated leucines are not conserved between mouse and human (Fig. S1, Table S4); p.L449P and p.G686D, which were already used for the calibration; and p.G1139S, which we routinely use as a “dead” control in the CIMRA assay (Fig. 1b, c). For 32 of the remaining 38 (84%) variants, CIMRA assay values were below 25% of wild-type activity (Fig. 3e) while four variants displayed 25–50% and two displayed >50% activity (Fig. 3e). Feeding these in vitro activities into our regression equation, followed by integration with the computational analysis–based Prior-P, resulted in the two-component classification of 35 of the 38 variants as class 4/5, whereas only 3 remained class 3. Importantly, none were falsely classified as class 1/2 (Table S5).
Calculating the sensitivities and specificities of the two-component classification procedure revealed that the sensitivity of classification to class 4/5 was 0.92 with a specificity of 1.00. The sensitivity of classification to class 1/2 was 0.78 with a specificity of 1.00.
Interlab CIMRA assay comparison
To independently investigate the reproducibility of the CIMRA assay, which is essential for its use as a diagnostic tool, MMR activity of ten MSH6 variants from the calibration set was determined in independent laboratories in Australia, the United States, and the Netherlands. Assay results appeared highly reproducible (Fig. 4).

MSH6 variants and controls were tested for CIMRA assay activity in different centers worldwide (see legend in figure). The MSH6 p.G1139S variant is included in every experiment as a repair-deficient control.12, 17 Bars represent mean ± S.E.M. of 3–4 experiments. Numbers below the diagrams indicate the International Agency for Research on Cancer (IARC) classification. LUMC Leiden University Medical Center, QIMR QIMR Berghofer Medical Research Institute, WT wild type.
DISCUSSION
The integration of multiple data sources to classify a variant has been widely endorsed by the variant classification community.4,5,8 By quantitatively combining results from computational analysis with those from the calibrated CIMRA we have developed, calibrated, and validated a two-component classification procedure.
To enable validation, we have used a genetic screen to generate an independent set of inactivating MSH6 variants. We have carefully validated the use of these variants as proxies for human pathogenic variants. We identified the biochemical mechanisms of MMR deficiency for nearly all, and found that 13 of the 43 inactivating substitutions identified in the genetic screen matched previously identified human MSH6 variants. Of these 13 variants, 2 had been assigned class 4/5, by using clinical criteria, whereas the others had remained unclassified. Thus, our small-scale genetic screen already enabled to assign pathogenicity to 11 human VUS demonstrating that, in addition to the two-component classification, genetic screens can be used as an independent tool to classify VUS. Even so, the genetic screen remains a surrogate for pathogenic human variants and a (small) possibility remains that not all of these screen-detected variants reflect true pathogenic human alleles. The sensitivities and specificities of our two-component procedure could therefore be (slightly) upwardly biased.
Independently testing the two-component classification on human counterparts of these “pathogenic” murine variants and on 18 class 1/2 human variants revealed that the two-component procedure classifies 88% of all MSH6 variants with very high sensitivities and specificities for both class 4/5 and class 1/2 variants. Importantly, no variant was misclassified (Tables S1, S3, S4). Combining results from our recent two-component classification procedure for variants in MLH1 and MSH215 with those for variants in MSH6 reveals that the discordance rate between clinical classification and the two-component classification is 1.4% at most (2 errors in 148 variants). The sensitivities and specificities of our two-component procedure compares favorably with other diagnostic tools used in clinical medicine.35 Based on these studies we surmise that the two-component procedure may greatly improve the classification of variants in MMR genes, specifically in MSH6. Furthermore, it is important to emphasize here that our observation that many “pathogenic” variants produced in the screen retain significant Msh6 expression (Fig. 3a) warrants great caution with the use of immunohistochemistry as a diagnostic criterion for LS.
With the increased deployment of exome sequencing for cancer susceptibility it is expected that not only the total number of identified variants, but also the relative incidence of variants with reduced penetrance in MMR genes will increase. The classification of intermediate-penetrance variants represents the next challenge. We anticipate that this will require the development of an extended classifier based on a Bayesian integration of quantified clinical criteria (including MSI and segregation) with our functional assay–based two-component classification.4 This classification may be aided by genetic screens as described here.
Finally, with the increased incidence of germline and somatic variants in MMR genes and in other cancer-predisposing genes, it is seminal to develop efficient and validated diagnostic tools to enable the translation of personalized genomics into personalized health care. The approaches followed here provide a template for the development of such tools.
References
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15:181–194.
Sijmons RH, Hofstra RMW. Clinical aspects of hereditary DNA mismatch repair gene mutations. DNA Repair (Amst). 2016;38:155–162.
Peltomäki P. Update on Lynch syndrome genomics. Fam Cancer. 2016;15:385–393.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Thompson BA, Spurdle AB, Plazzer J-P, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2013;46:107–115.
Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291.
Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66:464–472.
Rasmussen LJ, Heinen CD, Royer-Pokora B, et al. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat. 2012;33:1617–1625.
Rasmussen LJ. Approaches to diagnose DNA mismatch repair gene defects in cancer. DNA Repair (Amst). 2016;38:147–154.
Raevaara TE, Korhonen MK, Lohi H, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549.
Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007;67:4595–4604.
Drost M, Zonneveld JBM, van Hees S, Rasmussen LJ, Hofstra RMW, de Wind N. A rapid and cell-free assay to test the activity of Lynch syndrome-associated MSH2 and MSH6 missense variants. Hum Mutat. 2011;33:488–494.
Drost M, Zonneveld JBM, van Dijk L, et al. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat. 2010;31:247–253.
Drost M, Koppejan H, de Wind N. Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum Mutat. 2013;34:1477–1480.
Drost M, Tiersma Y, Thompson BA, et al. A functional assay–based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet Med. 2010;21:1486–1496.
Thompson BA, Greenblatt MS, Vallee MP, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2012;34:255–265.
Houlleberghs H, Goverde A, Lusseveld J, et al. Suspected Lynch syndrome associated MSH6 variants: a functional assay to determine their pathogenicity. PLoS Genet. 2017;13:e1006765.
Holmes J, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–5841.
Drost M, Lützen A, van Hees S, et al. Genetic screens to identify pathogenic gene variants in the common cancer predisposition Lynch syndrome. Proc Natl Acad Sci USA. 2013;110:9403–9408.
Goldgar DE, Easton DF, Byrnes GB, et al. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29:1265–1272.
Thompson BA, Goldgar DE, Paterson C, et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer Family Registry. Hum Mutat. 2012;34:200–209.
Hofstra RMW, Spurdle AB, Eccles D, et al. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum Mutat. 2008;29:1292–1303.
De Wind N, Dekker M, Claij N, et al. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet. 1999;23:359–362.
Swann PF, Waters TR, Moulton DC, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–1111.
Bacher JW, Abdel Megid WM, Kent-First MG, Halberg RB. Use of mononucleotide repeat markers for detection of microsatellite instability in mouse tumors. Mol Carcinog. 2005;44:285–292.
Yang G, Scherer SJ, Shell SS, et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150.
Tavtigian SV, Greenblatt MS, Harrison SM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–1060.
Mahmood K, Jung C-H, Philip G, et al. Variant effect prediction tools assessed using independent, functional assay–based datasets: implications for discovery and diagnostics. Hum Genomics. 2017;11:10.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087.e3.
Pearlman AH, Hsieh P. DNA mismatch repair and the DNA damage response. DNA Repair (Amst). 2016;38:94–101.
Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSα DNA lesion recognition complex. Mol Cell. 2007;26:579–592.
Dufner P, Marra G, Raschle M, Jiricny J. Mismatch recognition and DNA-dependent stimulation of the ATPase activity of hMutSalpha is abolished by a single mutation in the hMSH6 subunit. J Biol Chem. 2000;275:36550–36555.
Bowers J, Sokolsky T, Quach T, Alani E. A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. J Biol Chem. 1999;274:16115–16125.
Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3:255–261.
Vickers AJ. Decision analysis for the evaluation of diagnostic tests, prediction models and molecular markers. Am Stat. 2008;62:314–320.
Acknowledgements
We appreciate help from Alun Thomas (Huntsman Cancer Institute, Salt Lake City, UT) with the CIMRA assay calibration. This work was supported by the Dutch Digestive Foundation (grant FP 16-01) and the Dutch Cancer Society (grant UL 2013-5939).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Drost, M., Tiersma, Y., Glubb, D. et al. Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome. Genet Med 22, 847–856 (2020). https://doi.org/10.1038/s41436-019-0736-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0736-2
