
Abstract
Purpose
Kabuki syndrome (KS) (OMIM 147920 and 300867) is a rare genetic disorder characterized by specific facial features, intellectual disability, and various malformations. Immunopathological manifestations seem prevalent and increase the morbimortality. To assess the frequency and severity of the manifestations, we measured the prevalence of immunopathological manifestations as well as genotype–phenotype correlations in KS individuals from a registry.
Methods
Data were for 177 KS individuals with KDM6A or KMT2D pathogenic variants. Questionnaires to clinicians were used to assess the presence of immunodeficiency and autoimmune diseases both on a clinical and biological basis.
Results
Overall, 44.1% (78/177) and 58.2% (46/79) of KS individuals exhibited infection susceptibility and hypogammaglobulinemia, respectively; 13.6% (24/177) had autoimmune disease (AID; 25.6% [11/43] in adults), 5.6% (10/177) with ≥2 AID manifestations. The most frequent AID manifestations were immune thrombocytopenic purpura (7.3% [13/177]) and autoimmune hemolytic anemia (4.0% [7/177]). Among nonhematological manifestations, vitiligo was frequent. Immune thrombocytopenic purpura was frequent with missense versus other types of variants (p = 0.027).
Conclusion
The high prevalence of immunopathological manifestations in KS demonstrates the importance of systematic screening and efficient preventive management of these treatable and sometimes life-threatening conditions.
Similar content being viewed by others

INTRODUCTION
Kabuki syndrome (KS, OMIM 147920 and 300867) is a rare genetic disorder with an estimated prevalence of 1/32,000 people. KS is characterized by specific facial features, intellectual disability, skeletal anomalies, short stature, and dermatoglyphic anomalies, including persistence of fetal fingertip pads.1 KS is due to pathogenic variants in lysine-specific methyltransferase 2D (KMT2D, OMIM 602113)2 or lysine-specific demethylase 6A (KDM6A, OMIM 300128, an X-linked gene that escape partially to X inactivation).3 Those genes are part of the complex of proteins associated with Set1 (COMPASS), which is involved in chromatin remodeling by modifying epigenetic marks on histones.4 For 56% to 75% of KS individuals, the major gene involved is KMT2D.5 KDM6A is mutated in about 5% of KS individuals.6
Recurrent infections, mainly otitis media, abnormal immunoglobulin secretion,7 decreased memory cells, and poor vaccine response8 have been reported in isolated or small series of patients with KS but few authors have emphasized the potential severity of immune complication such as hypogammaglobulinemia and granulomatous–lymphocytic interstitial lung disease (GLILD).9 Autoimmune diseases (AIDs have also been reported; the most frequent manifestations include immune thrombocytopenic purpura10 with or without concurrent hemolytic anemia or autoimmune neutropenia,11 followed by vitiligo,12 autoimmune thyroiditis,13 diabetes type 1,14 Crohn disease,15 and membrane glomerulonephritis type 3.16 To our knowledge those immune manifestations have never been quantified by epidemiological tools. Actual guidelines for monitoring immune defects of KS are based on single-patient data or small series experience.
By taking advantage of a French collaborative survey on KS, we report a series of 177 individuals with KS due to KMT2D or KDM6A pathogenic variants; describe the immune phenotype, emphasizing differences by sex, age, gene, and type of pathogenic variant; and propose recommendations for immunologic survey in KS.
MATERIALS AND METHODS
Participants
We collected data for 177 individuals from a national registry (PHRC AOM-07-090 and additional individuals coordinated by Montpellier University Hospital; ClinicalTrials.gov identifier: NCT01314534). All patients gave their written informed consent before inclusion. Ethical approval was obtained from CPP2007-12-01, CPP Ile de France 2. All patients had a disease-causing variant in KMT2D or KDM6A.
This study relies in part on prospective data from the registry and retrospective data collected by a questionnaire addressed to 30 different French genetic centers that followed KS individuals from this registry, in coordination with the hematologist from the corresponding center. For subgroup analysis, individuals were divided into three age groups. From an endocrinological standpoint supporting hormonal changes during puberty, children were analyzed at <9 and 10 years old for females and males. Teenagers were analyzed at 9–17 and 10–18 years old for females and males, respectively, and adults at >17 and >18 years old for females and males, respectively.
Immunodeficiency
To assess immunodeficiency, data were extracted from medical records and the PHRC database. After data extraction, the clinician was asked by questionnaire if the individual had any recurrent or severe infections, especially ear, nose, and throat (ENT), pulmonary, or gastrointestinal infections. Recurrent infections are defined as three or more infections (sinusitis, otitis, bronchitis, pneumonia) in one year. Severe infections include those with failure to respond to oral antibiotics and/or the need for intravenous antibiotics or hospitalization. Susceptibility to infection was considered if the individual matched one of the criteria mentioned above. Biological values for complete blood count and quantitative serum immunoglobulin tests extracted from records were collected. A decrease in immunoglobulin levels of ≥2 standard deviations according to age was considered hypogammaglobulinemia. In cases of abnormal levels, we asked for results regarding immunoglobulin subclass, postimmunization antibodies, complement levels, and lymphocyte subset counts. Common variable immunodeficiency disorders (CVIDs) were defined by the combination of recurrent, severe, or unusual infections and hypogammaglobulinemia: a marked decreased (at least 2 standard deviations below the mean for age) in serum IgG and IgA.17
Autoimmunity
For autoimmune diseases, medical history of immune thrombocytopenic purpura (ITP), autoimmune hemolytic anemia (AIHA), thyroiditis, diabetes, vitiligo, etc. were specifically assessed. ITP was defined according to international criteria.18 AIHA was characterized by hemoglobin level <11 g/dL, a positive direct antiglobulin test, and at least one biological hemolysis criteria (reticulocyte count >120 × 109/L, haptoglobin level <10 mg/dL, or total bilirubin level >1 mg/dL). Vitiligo was considered if the individual had a depigmentation disorder of the skin without other etiology secondary to infection or treatment, for example. The diagnosis of autoimmune thyroiditis was based on increased level of antithyroid peroxidase (TPO) antibody (≥30 kU/l) or thyroglobulin (Tg).13
Variant screening and validation
Genomic DNA was isolated from peripheral EDTA-anticoagulated whole blood by using QIAamp DNA Blood Kits (Qiagen, Valencia, CA, USA). The entire coding sequence and intronic boundaries of KMT2D (NG_027827.1) and KDM6A (NG_016260.1) were screened for variants by Sanger sequencing of KMT2D according to routine diagnostic protocols or by next-generation sequencing (NGS) on a custom capture-based panel including KMT2D and KDM6A, then confirmed by Sanger sequencing. Obtained sequences were aligned against the hg19 (genome build: GRCh37) genome reference sequences from the National Center for Biotechnology Information (NCBI). Molecular results were analyzed by using the transcript NM_003482.3 for KMT2D and NM_021140.3 for KDM6A, Alamut Visual (Interactive Biosoftware, Rouen, France) and Ensembl website (http://grch37.ensembl.org/). Part of the genetic variants described in this registry were published previously.19 We consider a missense variant as likely pathogenic or pathogenic (Supplementary Table 3) when it arises de novo and we classified it as likely pathogenic or pathogenic on the VarSome website (https://varsome.com/).20
Statistical analysis
Descriptive statistics were reported: quantitative data are expressed with mean ± SD and range, and qualitative data by number (%). Statistical association between variables was tested by chi-square or Fisher's exact test otherwise (full data in Table 1). In our sample, females were older than males. Therefore, when necessary, we tested the confounding effect of these variables by multivariate logistic regression. Results of logistic regression are presented when they differ from results of univariate analysis.
RESULTS
Our series of 177 KS individuals included 89 males. Full data are available in Supplementary Table 2; 68 individuals were children (<9 or <10 years old) at the time of their last medical appointment; 66 were teenagers (<17 or <18 years old), and 43 were adults. The mean (SD) age at the time of the last medical appointment was 11.7 ± 10 (range 0–66) years. Five individuals died, three from immunopathological complications: acute bronchitis infection for two patients, hypovolemic shock consecutive to gastroenteritis and acute hemolysis at age 24 years due to chronic Evans syndrome for another. We identified KMT2D and KDM6A pathogenic variants in 162 and 15 KS individuals, respectively. For the type of pathogenic variant in KMT2D, we identified 67 nonsense, 50 frameshift, 35 missense, and 10 splice variants. For KDM6A, we identified 7 splice, 6 nonsense, 1 frameshift and 1 missense variants.
The prevalence of immunopathological manifestations in this registry is shown in Fig. 1, and the prevalence among subgroups is listed in Table 1. Overall, 78/177 (44.1%) individuals showed susceptibility to infections (Fig. 1). The most frequent infection was repeated ENT infection (47/177). Infections of the lower respiratory tract (6/177) and urinary tract (6/177) were less frequent. Bones, skin, gastrointestinal tract, cerebral, and ophthalmologic infections were occasionally reported. Two individuals had lung granulomas associated with hypogammaglobulinemia, for granulomatous–lymphocytic lung disease.

Prevalence of immunodeficiency and autoimmune disorders in Kabuki syndrome (KS). Presence denotes presence of one of the following items: AID autoimmune disease, AIHA autoimmune hemolytic anemia, HypoIg hypogammaglobinemia, IS infection susceptibility, ITP immune thrombocytopenic purpura, Oth other, Thy thyroiditis, Vit vitiligo. Absence denotes absence of the event. Not reported data were not available.
Repeated complete blood count was normal in all individuals from this registry. Quantitative serum immunoglobulin test was performed in 79/177 (44.6%) patients; 46/79 (58.2%) individuals had defects in secretion of IgA, IgM, or IgG Fig. 1). Combined hypogammaglobulinemia was observed in 28/79 (35.4%) individuals. CVID was observed in half (39/79, 49.4%), whereas 22/79 (27.8%) individuals were without hypogammaglobulinemia and infection susceptibility (Supplementary Table 2). Nonadequate protective postimmunization antibodies of antitetanic titers were found in 6/11 individuals with available data. We could not collect lymphocyte subset counts.
In total, 24/177 (13.6%) individuals had AID (Fig. 1); 10/177 (5.6%) had ≥2 autoimmune manifestations. ITP was the most frequent autoimmune complication (7.3%, 13/177); 12/13 (92%) individuals had the chronic form lasting more than 12 months. The second hematologic autoimmune manifestation was AIHA, including 7/177 (4.0%) individuals (Table 1); 6/7 individuals with AIHA had simultaneous or dissociated ITP (Evans syndrome). In total, 9/177 (5.1%) individuals had vitiligo (Fig. 1). Two adult females had autoimmune thyroiditis (1.1%). Finally, one individual had diabetes mellitus type 1, one had inflammatory bowel disease, one had antiphospholipid syndrome, and one had juvenile idiopathic arthritis (oligoarthritic form).
In total, 11/39 (28%) individuals with CVID had an AID as compared with only 2/22 (9%) without CIVD. The rate of AID was significantly increased for individuals with CVID (p < 0.1, Fisher’s exact test), whereas association between HypoIg and AID was not significant (p = 0.3).
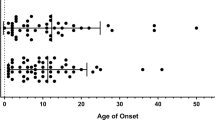
The prevalence of immunopathological manifestations was similar on subgroup analysis by age, gene (KMT2D or KDM6A), variant subtype, and sex (p > 0.05; Table 1). AID manifestations increased with age (p = 0.013, Table 1). Fewer than 4/68 (5.9%) children had AID manifestations as compared with 11/43 (25.6%) adults (Supplementary Fig. 2).
Genotype–phenotype correlations showed missense variants frequent in individuals with ITP (p = 0.027, Table 1). Also, autoimmune manifestations less frequently affected individuals with KDM6A than KMT2D variants, although not significantly (6.7% vs. 14.2%, p = 0.697; Table 1). The position of the pathogenic variants on the coding sequence was not associated with immune phenotype.
DISCUSSION
In this nationwide collaborative study, almost half of the 177 KS individuals analyzed had features of immunodeficiency and 13% had an AID. Prevalence of AID manifestations increased with age, with one quarter of adults showing at least one disease. Several authors had previously reported on CVID or autoimmunity in KS, but no large registry was studied. We focused on immunopathological manifestations in KS and described their prevalence, detailed features, and any genotype–phenotype correlations. We found that immunopathological manifestations were frequent and also seemed severe in KS.
Our findings may be understood by the loss of function of KMT2D and KDM6A. Indeed, KMT2D and KDM6A encode for proteins acting in a same biological complex called COMPASS. This complex allows for open-state chromatin by adding an activator trimethylation on the lysine 4 of histone 3 and removing the inhibitor trimethylation on the lysine 27.21 This situation translates epigenetically into a positive regulation of targeted gene transcription.22 KS individuals feature defective B-lymphocyte terminal differentiation, which probably explains the hypogammaglobulinemia. The literature describes significantly reduced number of total memory (CD27+) and class-switched memory B cells (IgM−) in Kabuki patients type 1 or without molecular study, compared with controls.23 The COMPASS complex helps in the transcription of the AID complex gene.24 This complex guides somatic hypermutation and class switch recombination, which is essential to B-cell differentiation, diversity, and efficiency.24 Pathogenic variants in KMT2D and KDM6A may lead to a dysfunction in this process and could explain the immune deficits observed in people with KS. The COMPASS complex is also involved in the epigenetic regulation of FOXP3,25 a key gene during the differentiation of naïve CD4+ T cells into T-regulatory cells, which contribute to the maintenance of peripheral tolerance.26 Pathogenic variants in KMT2D or KDM6A may lead to FOPX3 dysfunction and therefore a breakdown of T-cell tolerance, which could be the mechanism for the autoimmune phenotype observed in KS.
The strengths of our study are the large and multicentric setting and the retrospective collection of data that relied on genetics and hematoimmunolological rare disease networks in our country as well as the genotype–phenotype study. The wide distribution of age in our sample disallows conducting a reliable longitudinal analysis regarding autoimmune diseases. Several biological data regarding immunodeficiency were lacking. Some centers had different follow-up management; therefore, our missing data are mainly due to the intercenter management strategies. However, when data were available, data for 77.2% individuals were concordant for results for susceptibility to infection and hypogammaglobulinemia and thus we could correctly assess CVID in individuals (49.4% [39/79] with CVID and 27.8% [22/79] without hypogammaglobulinemia and infection susceptibility).27 In this series, only 79/177 (44.6%) individuals had a quantitative serum immunoglobulin test performed and registered in their medical record. This low percentage demonstrates that hypogammaglobinemia is not always tracked by clinicians in France. Therefore, immunoglobulin supplementation is not implemented or not early enough. This situation increases the risk of intercurrent infectious disease, leading to avoidable hospitalizations in these fragile individuals and fosters rare but critical conditions such as GLILD. However, most of the clinical manifestations secondary to immune impairment were mild to moderate, but for the five deaths in the registry, three were from immunological features and highlight the real need for adequate monitoring of these features in KS individuals.
We identify a high prevalence of immune manifestations in KS: 25.6% of our adult cohort had autoimmune manifestations as compared with 7.6–9.4% in the general adult population.28 The most frequent manifestations were isolated or combined autoimmune cytopenia, ITP, AIHA, or Evans syndrome). Prevalence of ITP usually decreases during childhood in the general population,29 but we observed the opposite in this series (p = 0.011; Table 1 and Supplementary Fig. 2). Also, 20% of general children with ITP have a chronic course lasting more than 12 months,30 whereas 92% of our KS sample had a chronic form, with second-line treatment.
One individual died from an acute hemolysis as a result of Evans syndrome. This severe situation is described in individuals with AIHA in the general population,31 more often in adults. Therefore, autoimmunity in KS should be carefully monitored and especially from pediatric to adult care.
Vitiligo was ten times more frequent in our series than in the general population.32 The sex ratio was 2 (6 males to 3 females) in our sample but 0.84 in the general population.33 We do not have an explanation for this discrepancy. The difference might not be significant because of the small number of individuals in this series.
We found thyroiditis in 2 (1.1%) adult females. In the literature, thyroiditis is usually described in adult females (94.5%).34 The prevalence of thyroiditis in the general population is 0.008%34 versus 1.1% in our registry. Here again, this data may be uncertain because of the small sample size.
On univariate analysis, women were more affected, but not significantly, by autoimmunity than were men, as observed in the general population.34
The rate of any autoimmune disease in KS individuals with CVID was 28% versus 9% without immune manifestations. This observation is related to the fact that CVID is frequently associated with AID in the literature,35 which led us to hypothesize that KS individuals with CVID should be supervised even more carefully for autoimmune manifestations. A larger sample of individuals is necessary to demonstrate an increased risk of AID in KS individuals with CVID.
For genotype–phenotype correlations, ITP was more frequent with missense than truncating variant carriage. However, we observed no difference of the distribution of missense pathogenic variants associated to ITP compared with other missense pathogenic variants at the exonic and protein level respectively (Supplementary Table 3 and Supplementary Fig. 3). Immune deficiency or autoimmune manifestations did not differ by type of gene (KMT2D or KDM6A). Autoimmune manifestations were fewer with KDM6A than KMT2D (6.7% vs. 14.2%), which is due to the younger median age of KDM6A than KMT2D individuals (8.1 vs. 13.7).
Patients with KS should have an assessment by an immunologist as soon as there is any doubt of dysimmunity, to avoid chronic diseases such as bronchiectasis or GLILD. The overall management of those patients should include clinical specific assessment, with a special attention to repeated ENT, bronchial, or digestive infection; lymphoproliferation; and symptoms of autoimmune diseases, mainly cytopenia or thyroiditis. We recommend checking by complete blood cell counts, IgG IgA IgM levels, T, B and NK lymphocytes phenotyping all patients at diagnosis. This blood work should be repeated it in case of recurrent or unusual infections. The immunization schedule should follow standard national guidelines. Measurement of T3, T4, and thyroid-stimulating hormone (TSH) levels is also indicated in case of clinical hypothyroidism. Patients with KS should have an assessment by an immunologist as soon as there is any suspicion of immunopathological manifestations, to avoid chronic diseases such as bronchiectasis or GLILD and to discuss specific treatments.
Prompt and adapted curative anti-infectious treatments, prophylactic antibiotics, and immunoglobulin substitution are to be considered on a case-by-case basis, based on guidelines available for patients with primary immunodeficiency.36
References
Niikawa N, Matsuura N, Fukushima Y, et al. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99:565–569.
Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793.
Lederer D, Grisart B, Digilio MC, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124.
Hu D, Gao X, Morgan MA, et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33:4745–4754.
Miyake N, Koshimizu E, Okamoto N, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet. 2013;161:2234–2243.
Banka S, Lederer D, Benoit V, et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin Genet. 2015;87:252–258.
Hoffman JD, Ciprero KL, Sullivan KE, et al. Immune abnormalities are a frequent manifestation of Kabuki syndrome. Am J Med Genet A. 2005;135A:278–281.
Lin J-L, Lee W-I, Huang J-L, et al. Immunologic assessment and KMT2D mutation detection in Kabuki syndrome: immunologic and genetic KMT2D analysis in Kabuki syndrome. Clin Gen. 2015;88:255–260.
De Dios JAA, Javaid AA, Ballesteros E, Metersky ML. An 18-year-old woman with Kabuki syndrome, immunoglobulin deficiency and granulomatous lymphocytic interstitial lung disease. Conn Med. 2012;76:15–18.
Ming JE, Russell KL, McDonald-McGinn DM, Zackai EH. Autoimmune disorders in Kabuki syndrome. Am J Med Genet. 2005;132A:260–262.
Almécija AC, Pérez V, Baro M, et al. Atypical autoimmune hematologic disorders in a patient with Kabuki syndrome. J Pediatr Hematol Oncol. 2019;41:e114–e115.
Zannolli R, Buoni S, Macucci F, et al. Kabuki syndrome with trichrome vitiligo, ectodermal defect and hypogammaglobulinemia A and G. Brain Dev. 2007;29:373–376.
Gürbüz F, Yüreğir ÖÖ, Ceylaner S, et al. Coexistence of Kabuki syndrome and autoimmune thyroiditis. J Clin Res Pediatr Endocrinol. 2016;8:105.
Fujishiro M, Ogihara T, Tsukuda K, et al. A case showing an association between type 1 diabetes mellitus and Kabuki syndrome. Diabetes Res Clin Pract. 2003;60:25–31.
Ho J, Fox D, Innes AM, et al. Kabuki syndrome and Crohn disease in a child with familial hypocalciuric hypercalcemia. J Pediatr Endocrinol Metab. 2010;23:975–979.
Nishizaki N, Fujinaga S, Hirano D, et al. Membranoproliferative glomerulonephritis type 3 associated with Kabuki syndrome. Clin Nephrol. 2014;81:369–373.
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190–197.
Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–2393.
Börgershausen N, Gatinois V, Riehmer V, et al. Mutation update for Kabuki syndrome genes KMT2D and KDM6A and further delineation of X‐linked Kabuki syndrome subtype 2. Hum Mutat. 2016;37:847–864.
Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35:1978–1980.
Lee MG, Villa R, Trojer P, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450.
Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661.
Lindsley AW, Saal HM, Burrow TA, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol. 2016;137:179–187.
Borchert GM, Holton NW, Edwards KA, et al. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS ONE. 2010;5:e11641.
Wei G, Wei L, Zhu J, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155.
Stagi S, Gulino AV, Lapi E, Rigante D. Epigenetic control of the immune system: a lesson from Kabuki syndrome. Immunol Res. 2016;64:345–359.
Westh L, Mogensen TH, Dalgaard LS, et al. Identification and characterization of a nationwide Danish adult common variable immunodeficiency cohort. Scand J Immunol. 2017;85:450–461.
Cooper GS, Bynum MLK, Somers EC. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 2009;33:197–207.
Moulis G, Palmaro A, Montastruc J-L, et al. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. 2014;124:3308–3315.
Grimaldi-Bensouda L, Nordon C, Leblanc T, et al. Childhood immune thrombocytopenia: a nationwide cohort study on condition management and outcomes. Pediatr Blood Cancer. 2017;64:e26389.
Aladjidi N, Leverger G, Leblanc T, et al. New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 2011;96:655.
Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51:1206–1212.
Zhang Y, Cai Y, Shi M, et al. The prevalence of vitiligo: a meta-analysis. PLoS ONE. 2016;11:e0163806.
Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243.
Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. 2012;3:210.
Mahlaoui N, Warnatz K, Jones A, et al. Advances in the care of primary immunodeficiencies (PIDs): from birth to adulthood. J Clin Immunol. 2017;37:452–460.
Hannibal MC, Buckingham KJ, Ng SB, et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155A:1511–1516.
Li Y, Bögershausen N, Alanay Y, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130:715–724.
Makrythanasis P, van Bon BW, Steehouwer M, et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: a genotype–phenotype study. Clin Genet. 2013;84:539–545.
Micale L, Augello B, Fusco C, et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6:38.
Acknowledgements
We thank the families as well as the French Kabuki Association (http://www.syndromekabuki.fr/) for their participation in this study. Part of this work was supported by the French Ministry of Health (Programme Hospitalier de Recherche Clinique national, AOM 07-090), Fondation Maladies Rares, and the French Kabuki Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
David Geneviève is a consultant for the Takeda Society. The society did not have any influence on the content of this report or its publication. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Margot, H., Boursier, G., Duflos, C. et al. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genet Med 22, 181–188 (2020). https://doi.org/10.1038/s41436-019-0623-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0623-x
Keywords
This article is cited by
-
Five years of experience in the Epigenetics and Chromatin Clinic: what have we learned and where do we go from here?
Human Genetics (2024)
-
Epigenetic regulation of pediatric and neonatal immune responses
Pediatric Research (2022)
