
Abstract
The National Institutes of Health (NIH) established the Rare Diseases Clinical Research Network to address the unique challenges of performing research on rare diseases. The Urea Cycle Disorders Consortium (UCDC) was one of the original ten consortia established. The UCDC represents a unique partnership among clinicians, patients, and the NIH with a primary goal of increasing the development of therapeutics that improve patient outcomes for persons affected with a UCD. Based in part on financial incentives associated with the Orphan Drug Act biopharmaceutical and investment entities have an intense interest in engaging with research consortia like the UCDC, which have compiled potentially valuable longitudinal data characterizing outcomes in a relatively large number of affected individuals. We describe the UCDC experience and the bases for evaluating partnerships with such private entities. We review early industry interactions, the development of policies and procedures, and describe the establishment of an Industry Relations Committee, including guiding principles. Challenges encountered, particularly in the transition when products are approved, and potential solutions are discussed. By building a framework for industry partnerships that guides us in resolving inevitable challenges, we can enthusiastically pursue novel and promising collaborations that can lead to breakthroughs in therapeutic interventions for patients.
Similar content being viewed by others

INTRODUCTION TO THE RARE DISEASE CONSORTIUM NETWORK AND THE UCDC: INDUSTRY RELATIONSHIPS
In response to the Rare Diseases Act of 2002, the National Institutes of Health (NIH) established the Rare Diseases Clinical Research Network (RDCRN) to address the unique challenges of performing research on rare diseases.1 This was undertaken using a cooperative agreement mechanism, firmly establishing the partnership and collaboration of NIH with the scientific community in initiating these research efforts. The vision of the RDCRN has been to bring together clinical research teams in partnership with NIH and patient advocacy groups to advance the understanding of rare diseases and to accelerate the development of treatments for these conditions, and to provide a resource for the rare diseases community.2 In addition to the inclusion of families as key partners, collaborations with industry serve to promote the development of new therapies. The Urea Cycle Disorders Consortium (UCDC), a partnership between the National Urea Cycle Disorders Foundation, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the Office of Rare Diseases Research at the National Center on Advancing Translational Sciences (NCATS), was one of the original ten consortia established and awarded support in the initial RDCRN funding cycle.3 Since its founding, the RDCRN has grown to 21 consortia with 98 patient advocacy group partners. The UCDC itself, currently in its third 5-year funding cycle, has expanded to include 16 clinical research sites in the United States, Canada, and Europe. The UCDC has embraced an international collaborative approach to UCD research, fostering 38 studies, including the Longitudinal Study of Urea Cycle Disorders with more than 700 participants enrolled.
The UCDC represents a unique partnership among clinicians, patients, and the NIH with a primary goal of increasing the development of therapeutics that improve patient outcomes for persons affected with a UCD. Patients with rare diseases, and the patient advocacy organizations that represent them, play integral roles in supporting biomedical research. In the United States, the Orphan Drug Act of 1983 was championed by families with rare diseases led by Abbey Meyers, a mother of a child with Tourette syndrome and the first president of the National Organization for Rare Disorders (NORD).4 In the United States, a rare disease is defined as one that affects fewer than 200,000 people;5 the goal in promotion of the Orphan Drug Act was to facilitate advancement of therapies for rare diseases, termed “orphan” drugs. Orphan drugs are developed to treat conditions meeting the definition of a rare disease. Patient advocacy groups have contributed to research endeavors through various mechanisms such as aiding in patient recruitment, providing support for families, as well as financial support for research and drug development.6,7,8 Research directly supported by patient advocacy groups has resulted in the development of new therapies for disorders such as cystic fibrosis and Pompe disease.
These opportunities for collaboration fit one of the stated goals of the legislation that established the RDCRN—to bring orphan products more rapidly to market to improve the care of individuals with rare disorders in the United States. In an effort to promote this goal, the UCDC developed a number of collaborations with the pharmaceutical industry and biotech firms to accelerate development of orphan products and diagnostic technology for UCD.
GROWING INTEREST IN RARE DISEASES BY PHARMA AND BIOTECH INDUSTRY
Developing new treatments for rare diseases is complicated by the difficulty in recruiting a sufficiently large population of patients for study from the small number of affected individuals. The evidence base in rare diseases is therefore limited. Prior to creation of the UCDC and the Longitudinal Study of UCDs, our knowledge of the natural history of these inborn errors of metabolism derived from observations in small case studies rather than a large, prospectively studied cohort. A paucity of enrollees makes the assessment of causal relationships difficult and subject to bias from the inability to ensure comparability. It may also be difficult to discern generalizable results as the cohort under study may not be representative of the condition as a whole. The detection of genotype–phenotype correlations is often hindered by the absence of a variant that prevails in a sufficiently large patient subgroup. Therapeutic trials lack statistical robustness because of small sample sizes.9
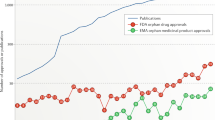
Despite these inherent difficulties in rare disease research, development of translational therapeutics, specifically those targeting rare genetic diseases, continues to increase. This is due, in large part, to legislative incentives that have stimulated considerable interest in rare diseases on the part of private capital. Under the Orphan Drug Act, incentives for orphan designation include tax credits for clinical trial–based research and development expenditures, waivers of the New Drug Application filing fee (approximately $2.4 million in FY2016) and a seven-year market exclusivity.10 Between 2001 and 2010 the US Food and Drug Administration (FDA) approved 62 novel therapeutics designated as orphan products, representing 27.9% of all approved novel therapeutics.11,12 During a similar time period the European Medicines Agency (EMA)13 considered 605 therapeutics in various stages of development with the orphan designation. As a result, in 2016 the global orphan drug market was estimated to be worth $114 billion. Worldwide orphan drug sales are forecasted to total $209 billion by 2022, representing 21.4% of prescriptions exclusive of generics.14 The compound annual growth rate of the orphan drug market between 2001 and 2010 was 25.8%, compared with only 20.1% for a matched control group of nonorphan drugs.15
The average cost of a phase III clinical trial for an orphan drug is $103 million, about half that of nonorphan drugs at $193 million.16 The Orphan Drug Act additionally provides a 50% US tax credit, thereby effectively reducing the cost of an orphan drug trial to approximately $51 million.16 The median patient cohort for a phase III orphan drug trial is 538 patients compared with a median 1558 patients for nonorphan drugs.16 According to published analyses, there is no statistically significant difference in trial length for orphan versus nonorphan drugs, which averaged 2.88 years.16 Due to FDA priority reviews for orphan drugs, FDA approval times averaged 10 months for orphan drugs versus 13 months for nonorphan drugs.16 The average return on investment for an orphan drug was 1.14 times greater than a nonorphan drug.16
Despite the intent to improve treatment for rare diseases, there has been growing concern about misuse of the Orphan Drug Act.17,18,19 In 2017, three US senators sent a letter to the US Government Accountability Office (GAO) to question whether the Orphan Drug Act was resulting in the development of rare disease therapeutics as intended.20 Whether or not pharmaceutical companies are attempting to misuse the Orphan Drug Act, it is clear that the Orphan Drug Act alone was not a panacea for all rare diseases. The majority of FDA approved orphan drugs focus on similar diseases with cancer (31.9%) and infectious diseases (8.3%) therapeutics being the most common21 and less than 30% of orphan drugs targeted pediatric diseases.22 Despite these flaws, orphan drug designations have resulted in increased interest in rare disease therapeutics. Orphan drug designations have been granted or approved for rare diseases including 122 rare diseases in the EU and 300 rare conditions in the United States.23 In the EU it was recently estimated that over 450 therapies were in development for rare disease in large part due to various orphan drug protections.11,12 It is clear that an unmet therapeutic need for a number of rare diseases still exists. The financial incentives associated with the Orphan Drug Act alone are not enough to ensure the development of new therapeutics for rare diseases. This highlights the importance of continued collaboration between rare disease advocates and biopharmaceutical companies.
As a result, biopharmaceutical and investment entities have an intense interest in engaging with research consortia like the Rare Disease Clinical Research Network (RDCRN), which have compiled potentially valuable longitudinal data characterizing outcomes in a relatively large number of affected individuals. Longitudinal studies can determine frequency and variability of rare conditions and define what comorbid conditions exist. RDCRN investigators are sought after because they can serve as experts regarding the rare diseases under study in clinical trials and have access to longitudinal study participants as potential subjects for research trials. The RDCRN consortia also accumulate a rich data resource on a large pool of patients available to participate in studies. These subjects are typically on more homogeneous, state-of-the-art treatment protocols than is common for other rare disorders. The challenges associated with rare disease research and the economic incentives to develop rare disease therapeutics have resulted in an increasing number of academic and industry partnerships.24,25
The UCDC represents a unique partnership between clinicians and patients with a primary goal of increasing the development of therapeutics that improve patient outcomes. To achieve this objective, engaging with industry to promote drug development and defining the roles and boundaries of these relationships have become increasingly important. Herein we describe the UCDC experience and the guiding principles that we developed for evaluating partnerships with such private entities.
EARLY INDUSTRY INTERACTIONS
The UCDC has stimulated considerable interest from pharmaceutical companies that seek to develop novel therapeutics, including enhanced ammonia-scavenging therapies, gene replacement therapy, and hepatocyte-based therapy. In the latter two instances the goal is to correct the underlying congenital enzymatic deficiency that results in a failure of ureagenesis and consequent hyperammonemia and neurologic injury. Many of these pharmaceutical companies have approached the UCDC seeking the collective expertise of the UCDC investigators and access to the unique data that the consortium have compiled over the past 11 years, particularly from the 700+ participant longitudinal study data set. Longitudinal studies can determine the frequency and variability of rare conditions and define relationships to comorbid conditions, as well as risk factors that develop over time, data critical to the initiation of drug trials. To avoid the costs and lengthy process of conducting their own background research to inform the design of clinical trials and/or postmarketing surveillance, they have solicited the collaboration of the UCDC.
Many of the companies developing products for rare diseases are small startups that are ultimately acquired by larger companies after their product receives FDA approval. Although the opportunity for postmarket profits can be significant, initial financial resources are typically limited. Venture capital companies contemplate whether to invest in these startups and UCDC investigators receive many requests from companies and capital research firms seeking information on future market potential.
DEVELOPMENT OF POLICIES AND PROCEDURES
An anonymized raw data set from all RDCRN studies is available via the public database of Genotypes and Phenotypes (dbGaP) 3–5 years after data are collected, however the therapeutics industry’s development timeline typically requires more expeditious access to curated data. When it became apparent that developing collaborations with industry were going to require review and structure, the UCDC leadership worked with a technology transfer advisor and intellectual property attorney at Children’s National Health System (UCDC’s lead institution). The UCDC developed a “master services agreement” that included a definition of the use of the data for a specific purpose, with clearly defined limits for how the data may be used, shared, or distributed; UCDC and company rights to data; confidentiality, publication rights, and intellectual property; as well as a scope of work and timeline. Hourly rates were established for work performed by the UCDC investigators for projects on behalf of collaborations with industry partners. Over the course of the consortium’s efforts our own insight and understanding of the issues has deepened and molded our responses. The same is true of our NIH partners, leading to an ongoing effort to provide a guidance for principles for third-party collaboration for RDCRN consortia.
A critical insight is the recognition that the data accumulated by the UCDC have intrinsic value. The implications of this are still being explored, and the insight has not yet been fully developed into a definitive action plan that is consistent with the organizing principles and administrative processes of all the UCDC partners, including the NICHD. It is important to be clear that the costs of development of the UCDC infrastructure and data collection have already been borne by the agencies that fund the UCDC (NIH and philanthropic sources). The cost for retrieval and curation of the data for activities outside of the aims of the cooperative agreement remains a point for discussion with those seeking such access.
The consortium recognized that meeting industry data requests required effort over and above that needed to address predefined research aims and thus responded by creating a reimbursement structure to cover these added costs. The UCDC, working with the Data Management and Coordinating Center (DMCC), developed a complete list of hourly rates for these activities that is provided to industry collaborators before work begins. An estimate of the hours expected based on the scope of work is prepared and agreed upon, with the expectation that overages will be approved by the industry collaborator before work is done. A review of work effort indicates that a baseline effort of 1–5% by administrative and DMCC staff was provided for industry work over the last year. During intermittent intense interactions in collaborations with industry, up to 20% time could be required in a period of time where work was being concentrated. The creation of a reimbursement structure that includes data curation ensures that public funds are allocated as intended and that industry assumes responsibility for activities that address its particular interests. The landscape of research–industry collaboration will continue to evolve in the setting where clinicians who care for those affected with rare diseases are also likely to be the very experts needed to safely conduct clinical trials and to be members of an a rare disease consortium like the UCDC.
One of the challenges faced is that there is no corporate structure for the consortium to facilitate significant business activities, nor is that consistent with the mission of the consortium. The result is that the lead institution, which provides the financial infrastructure for the consortium (in the case of the UCDC, Children’s National Health System) provides the necessary functional business support. Ultimately, there is inherent conflict in joint activities between academic institutions and investigators, NIH, and industry; namely, that while there is a shared agenda to achieve some common good, the underlying drivers of decision-making are substantially different between the partners.
One company engages the UCDC to conduct a postmarketing surveillance protocol, which includes an annual report, to meet the company’s FDA annual reporting requirements. Participants consent to participate in this study for postmarketing surveillance purposes and agree to share a limited data set with the company through the annual report. This study requires only a few case report forms, in addition to the data already collected for the UCDC longitudinal study. Although this study involves modest additional effort for the site principal investigators (PIs) and coordinators, the operations to assist in fulfilling this responsibility for the company have required more time than is actually billed, including tracking and invoicing for participant visits, routing payments to the participating sites for conducting visits and institutional review board (IRB) activities, ensuring that the contract with our data management center is updated annually, and payment to the data management center for its work in preparing the annual report. Future contracts must ensure that all additional effort, both administrative and that related to collection and processing of extra information needed to satisfy an industry request or project, must be fully compensated. Ultimately, there have been costs to the consortium for industry interactions that were borne as necessary to the process of improving therapies for UCD patients. As work beyond the scope of the cooperative agreement, institutional indirect costs for the host of the consortium may need to be adjusted to reflect the cost for increased administrative needs.
ESTABLISHING AN INDUSTRY RELATIONS COMMITTEE: GUIDING PRINCIPLES
To manage these interactions and relationships, the UCDC established an Industry Relations Committee (IRC) in 2015. This group is constituted of members of the consortium who assist in developing policies for the consortium regarding industry relations and who can serve as a peer group in the management of conflicts of interest as necessary. Patient and family interests are assured by including a member representing the National Urea Cycle Disorders Foundation (NUCDF), and NIH perspectives are represented by inclusion of an IRC member from a funding institute. The committee has reviewed and revised the UCDC’s data use and publication policy sections relating to industry and has developed a new policy and supporting procedures to guide interactions with industry to which all of the members of the UCDC executive committee have agreed (see Supplementary Information).
The goals of the IRC are to promote transparency, clarify the mutual responsibilities of UCDC investigators and potential partners in the private sector, and to ensure that all agreements between industry and the UCDC safeguard patient interests and conform to existing NIH standards with regard to the conduct of clinical research. To this end, the IRC sought to develop explicit criteria that would provide UCDC investigators with guidelines for fostering relationships that avoid conflicts of interest and maintain the confidence of both the patients and families who participate in the longitudinal study and the public who fund UCDC efforts through NIH support. Central to these goals is an emphasis on transparency, honesty, and an acknowledgment that even the perception of a conflict of interest must be disclosed and managed, if necessary. The IRC developed principles that govern UCDC–industry interactions (Table 1).
Individual investigators who are members of the UCDC are free to participate in independent industry collaborations as individuals outside the UCDC, but they agree to disclose information about the nature of this involvement with the other members of the consortium, including NICHD.
CHALLENGES ENCOUNTERED: DEVELOPING SOLUTIONS
Creation of the RDCRN reflected the hope that its member consortia would both advance understanding of rare diseases and develop therapeutics to improve patient outcomes. The strength of the RDCRN consortia infrastructure is attracting pharmaceutical companies interested in rare disease drug development. Fostering productive relationships with partners in the private sector raises challenges. From the UCDC perspective, understanding the fundamental differences between the goals of industry and those of a research consortium is a paramount requirement to begin developing solutions to these challenges. While the hope is that both parties prioritize patient interests, the reality is that decisions made by those in industry fundamentally acknowledge the legitimate interests of shareholders while the UCDC is vested in its commitments to NIH and patient advocacy organization partners, and to upholding its guiding principles related to patient protections and the interests of patients. Reconciling these priorities is challenging, especially when the conduct of research becomes interdependent upon private and governmental support. In the examples to follow, we share what we have learned from the UCDC's experiences over the last 16 years.
The potential for conflicts of interest creates challenges. Although a conflict of interest (COI) can arise in many forms, such as academic self-interest, financial COIs are a particularly salient issue when discussing the relationship between academia and industry. It is important to note that increased scrutiny was placed on a financial COI following the failed research trial of a vector-administered gene therapy trial for ornithine transcarbamylase deficiency (OTCD) that led to the death of a research subject in 1999.26 Although the financial COI in the initial OTCD gene therapy trial may have been overstated in the popular press,27 the UCDC has emphasized the importance of mitigating possible COI in recognition of the importance of both the reality, and the public perception, of any potential COI.
As a member of a research consortium such as the UCDC, an investigator has professional expertise that is valuable to private industry. Involvement with the consortium inherently increases the value of the individual investigator, whose knowledge base is amplified by interactions with colleagues and by access to the data that the consortium collects. Conversely, the value and objectives of the consortium are enhanced by the creative participation of each of its researchers. However, in the interest of transparency, the inherent conflicts, both intellectual and financial, need to be recognized, reported, and managed.
Confidentiality agreements are a necessary element from the perspective of industry, but pose an additional challenge for investigators. The success of a research consortium presupposes an atmosphere that favors open scientific dialogue. As members of the consortium become increasingly valuable as consultants to industry, individual investigators may find that they have committed to competing confidentiality agreements that could be in conflict with one another. Complete avoidance of such conflicts may not be not feasible, but consortium members must agree to (1) transparency in disclosing these arrangements, and (2) acknowledgment of the primacy of the scientific and patient-oriented goals of the consortium and the need to preserve these aims ahead of any goals of industry. The consortium leadership and NIH are arbiters of conflicts identified by investigators or the IRC that an individual researcher is unable to resolve through negotiation.
Another challenge and potential source of conflict emerges when different pharmaceutical companies seek to perform multiple clinical trials that must compete for the scarce numbers of patients with a rare disorder such as a UCD. The consortium recognizes the right of patients and families to participate in studies that may offer benefit, but the consortium also appreciates the stress families may endure if they are solicited by competing pharmaceutical firms. We also recognize the need to provide an informed evaluation of available trials for decision support for participants but do not have a mechanism by which such an evaluation could be generated. Moreover, the UCDC needs to maintain an open view of trials that could improve care for affected families and is not in a position to endorse a given product as a consortium. Rather, the individual expert investigators who participate in the UCDC must make decisions about engagement in trials based on the clinical needs of their patients. The NUCDF provides educational resources to patients and their families to assist them in making informed decisions about participation in clinical trials. This continues to be an issue for which there is no obvious solution.
The UCDC has asked its members to provide information about their industry relationships. Recently, the IRC elected to provide access to this information on a real-time basis to all members of the consortium through use of a shared spreadsheet (the Conflict Management Form) that is available to all consortium members, leadership, and administrative staff. Members are requested to update the form with any new relationship(s) and, at a minimum, to provide updated details on an annual basis. While the UCDC does not restrict the rights of consortium members to participate outside the consortium in activities with industry partners, it does seek to ensure that those involvements are transparent so that colleagues can judge for themselves the impact this may have on mutual decision-making.
PUTTING CONTACTS WITH INDUSTRY INTO OPERATION: CONTRACTS
The value of the consortium to industry partners goes beyond investigator expertise and providing access to potential participants in research projects. A consortium such as the UCDC has value with regard to its extensive data set, which has been painstakingly assembled and curated by the members of the consortium and their support teams. The longitudinal study data set represents a nearly irreplaceable resource regarding the natural history of these conditions. This kind of information is essential to the development of new therapeutic agents and their utility in the UCD patient community.
This value of the consortium includes both the data in the longitudinal study of UCD and the collective expertise of the site investigators and leadership team, which has been accumulated with federal funding, and must be appropriately safeguarded, supported, and sustained. Specific intellectual property issues that are considered in developing contracts with industry are described in Table 2.
THE PRICE OF SUCCESS
As more companies invest in rare disease therapies, the potential exists for significant competition for a limited number of qualified clinical trial subjects, especially since a common requirement for enrollment in a therapeutic trial is that the patient be “naïve” with regard to participation in any other trial. The Consortium does not covet the role of a “court” that decides which therapeutic trial is most meritorious. Obviously, the public interest is ill-served if trial enrollment is determined not by the therapeutic potential of a putative agent, but by the company that is in a position to provide the most remuneration to researchers and enrollees, or even by the company that is able to be “first to clinical trial” even though the product may not represent the best therapeutic option.
It is also the expectation of UCDC that industry partners should be respectful of the need for continuing research in the rare disease setting and that postmarketing activities for new agents should not undermine the goals and principles of sound science and patient protection. UCDC industry partners must acknowledge that the UCDC represents the interests and needs of a community of affected patients and their families, not an agglomeration of individual subjects. Further, any cooperation by the UCDC with the private sector is contingent on scrupulous adherence to all NIH requirements for protection of human subjects and data sharing, as well as FDA regulations with regard to safeguarding patient safety and privacy (protected health information) during conduct of clinical trials and following FDA approval of a new medication. Data shared with industry collaborators is always de-identified unless the participant has consented to share his or her data for a specific purpose.
Our experience to date highlights the immense financial reward to be realized by the successful development of new therapeutics for rare diseases. Realizing a great profit generally obliges the assumption on the part of investors of high risk as well as prodigious investment to bring a new drug to market. Obviously, it follows for any rare disease that the cost per patient will be quite high when the pool of prospective users is relatively small. Postapproval, the costs of aggressive marketing and salesforces bloat the drug price. It is difficult to discern when a fair compensation for a large investment becomes predatory pricing, with a markup for medications out of proportion to recovery and reasonable profit following investment, particularly when the development of the drug was dependent upon resources like the UCDC that are subsidized by NIH and ultimately the taxpayers. A rational balance must be established between the legitimate needs of investors and those of the patient community. It is the position of the UCDC that, whenever feasible, this putative tension will be resolved by assigning primacy to the well-being of the patient community, with special emphasis on ensuring access to the new medication by patients whose access may otherwise be limited due to cost or lack of requisite insurance coverage.
The high cost of the new therapeutic agents continues to engender concern among members of the Consortium and the families of affected patients. We are united in our commitment to continue to aggressively seek opportunities for advancing therapy for these rare diseases, but remain guarded in our acceptance of the rationale for very high drug prices. Premium pricing for orphan drugs has raised significant and legitimate questions for public debate, as illustrated by the recent call for investigations into the “gaming” of the Orphan Drug Act and allegations that patient assistance programs enable price-fixing. In March 2017, the GAO opened an investigation on behalf of Congress into skyrocketing orphan drug pricing.28 The UCDC and UCD patients and families alike oppose predatory pricing, monopolies, and withholding of medications, particularly when (a) patients lack an equivalent therapeutic alternative, and (b) development of the medication would have been impossible without the active cooperation and support of the NUCDF and the patient community, including participation of patients in pivotal clinical trials. Neither is it appropriate to transfer high drug costs to third-party payers. These issues include new agents, the provision of drugs with expired orphan drug exclusivity, and those no longer on patent, and place patient care and outcomes at risk.29
The potential for disagreement always exists between the Consortium and putative partners in industry—no matter how diligently either side tries to avert conflict. An important lesson has been that the research community, the advocacy group, and industry partners often work well together during the development phase of a new therapeutic, when all agendas are aligned on getting a better treatment into clinical use. After FDA approval, though, the agenda of the industry partner diverges from that of the academic researchers and the advocates, making the interactions more complex. A company is responsible to its stockholders and may adopt tactics that are not favorable for the scientific collaboration anticipated by the consortium members. Examples include engaging subjects in trials that might limit participation in core UCDC studies or adopting marketing tactics that have potential to misrepresent the relationship of the company with the UCDC. Such activities must necessarily result in strong response by the UCDC to protect its integrity and scientific interests, the privacy and confidentiality of engaged research subjects, and to safeguard the trust that the NIH and the patient community have placed in us.
CONCLUSION
Accelerating the development of therapeutics that improve outcomes for patients with rare diseases presupposes constructive dialogue and cooperation between the research consortia and potential partners in the private sector. Indeed, the promise of collaborative partnerships and the role of the Rare Diseases Clinical Research Network consortia as facilitators of research for patients with rare disorders is beginning to bear fruit. An increasing number of therapeutic agents are in the pipeline and coming to market. Careful stewardship and transparent ongoing discussions between the RDCRN stakeholders—the research teams, NIH, and the communities of patients and families with rare disorders—will continue to ensure safeguards for patient privacy and productive use of information gained from the gift of their participation in research.
Patients and their families with urea cycle disorders have put their trust in the integrity and scientific expertise of the UCDC to advance research and development of therapeutics that improve outcomes, without exploiting their vulnerabilities. By building a framework for industry partnerships that guides us in resolving inevitable challenges, we can enthusiastically pursue novel and promising collaborations that can lead to breakthroughs in therapeutic interventions for patients. We hope that our lessons learned will inform and benefit other rare disease research consortia, patient advocacy organizations, industry collaborators, and, overarching, the interests of affected patients and families.
References
Shimkus J. H.R.4013–107th Congress (2001–2002): Rare Diseases Act of 2002. 6 November 2002. https://www.congress.gov/bill/107th-congress/house-bill/4013. Accessed 3 October 2017.
Batshaw ML, Groft SC, Krischer JP. Research into rare diseases of childhood. JAMA. 2014;311:1729–1730.
Seminara J, Tuchman M, Krivitzky L, et al. Establishing a consortium for the study of rare diseases: the Urea Cycle Disorders Consortium. Mol Genet Metab. 2010;100 suppl 1:S97–105.
Wikipedia. National Organization for Rare Disorders. 2017. https://en.wikipedia.org/w/index.php?title=National_Organization_for_Rare_Disorders&oldid=797804566.
National Center for Advancing Translational Sciences–Genetic and Rare Diseases Information Center. Diseases. https://rarediseases.info.nih.gov/diseases. Accessed 12 April 2019.
Griggs RC, Batshaw M, Dunkle M, et al. Clinical research for rare disease: opportunities, challenges, and solutions. Mol Genet Metab. 2009;96:20–26.
Panofsky A. Generating sociability to drive science: patient advocacy organizations and genetics research. Soc Stud Sci. 2011;41:31–57.
Koay PP, Sharp RR. The role of patient advocacy organizations in shaping genomic science. Annu Rev Genomics Hum Genet. 2013;14:579–595.
Fishman MC. Power of rare diseases: found in translation. Sci Transl Med. 2013;5:201ps111.
Federal Register. Orphan Drug Regulations. 19 October 2011. https://www.federalregister.gov/documents/2011/10/19/2011-27037/orphan-drug-regulations. Accessed 16 May 2017.
Hoffman EP, Workshop Participants, TREAT-NMD Alliance. Facilitating orphan drug development: proceedings of the TREAT-NMD International Conference, December 2015, Washington, DC, USA. Neuromuscul Disord. 2017;27:693–701.
Downing NS, Shah ND, Aminawung JA, et al. Postmarket safety events among novel therapeutics approved by the US Food and Drug Administration between 2001 and 2010. JAMA. 2017;317:1854–1863.
Morel T, Lhoir A, Picavet E, et al. Regulatory watch: the orphan drug pipeline in Europe. Nat Rev Drug Discov. 2016;15:376.
Report | EvaluatePharma Orphan Drug 2017 | Evaluate. http://info.evaluategroup.com/rs/607-YGS-364/images/EPOD17.pdf. Accessed 3 November 2017.
Silverman E. Orphan drugs: “rare” opportunities to make money. Forbes. https://www.forbes.com/sites/edsilverman/2012/08/23/orphan-drugs-rare-opportunities-to-make-money/. Accessed 4 October 2017.
Evaluate. EvaluatePharma Orphan Drug 2015 Report: worldwide orphan drug sales forecast to total $178bn. https://info.evaluategroup.com/od2015-lp.html. Accessed 3 November 2017.
Tribble SJ. FDA moves to rein in drugmakers’ abuse of orphan drug law. https://www.npr.org/sections/health-shots/2017/09/13/550700062/fda-moves-to-rein-in-drugmakers-abuse-of-orphan-drug-law. Accessed 12 April 2019.
Tribble SJ. FDA moves to guard against abuse of ‘orphan drug’ program. Kais Health News. September 2017. https://khn.org/news/fda-moves-to-guard-against-abuse-of-orphan-drug-program/. Accessed 12 April 2019.
Sullivan T. Senators ask GAO to investigate potential Orphan Drug Act abuse. https://www.policymed.com/2017/04/senators-ask-gao-to-investigate-potential-orphan-drug-act-abuse.html. Accessed 12 April 2019.
Tribble SJ, Lupkin S. Three key senators ask GAO to investigate possible abuses of the Orphan Drug Act. Kais Health News. March 2017. https://khn.org/news/three-key-senators-ask-gao-to-investigate-possible-abuses-of-the-orphan-drug-act/. Accessed 12 April 2019.
Rodriguez-Monguio R, Spargo T, Seoane-Vazquez E. Ethical imperatives of timely access to orphan drugs: is possible to reconcile economic incentives and patients’ health needs? Orphanet J Rare Dis. 2017;12:1.
Thorat C, Xu K, Freeman SN, et al. What the Orphan Drug Act has done lately for children with rare diseases: a 10-year analysis. Pediatrics. 2012;129:516–521.
Giannuzzi V, Conte R, Landi A, et al. Orphan medicinal products in Europe and United States to cover needs of patients with rare diseases: an increased common effort is to be foreseen. Orphanet J Rare Dis. 2017;12:64.
Coles LD, Cloyd JC. The role of academic institutions in the development of drugs for rare and neglected diseases. Clin Pharmacol Ther. 2012;92:193–202.
Mavilio F. Developing gene and cell therapies for rare diseases: an opportunity for synergy between academia and industry. Gene Ther. 2017;24:590–592.
Wilson JM. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96:151–157.
Rosenbaum L. Beyond moral outrage—weighing the trade-offs of COI regulation. N Engl J Med. 2015;372:2064–2068.
Rademacher K, Lima HA. Orphan drugs: understanding the payer perspective. J Clin Pathw. 2017;3:29–30.
Tribble SJ, Lupkin S. Drugmakers manipulate orphan drug rules to create prized monopolies. Kais Health News. January 2017. https://khn.org/news/drugmakers-manipulate-orphan-drug-rules-to-create-prized-monopolies/. Accessed 3 November 2017.
Acknowledgements
The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The Urea Cycle Disorders Consortium is also supported by the O’Malley Foundation and the Kettering Fund. We would specifically like to acknowledge feedback from NICHD program officer Danuta Krotoski and science officer Tracy King.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The following conflicts of interest (2017–present) are reported. S.A.B.: advisory board, Horizon Pharma; PI Horizon Pharma: HPN-100-009 Protocol: An Open Label Study under Two Years of Age; C.R.C.: site co-PI Orphan Europe/Recordati NCGC Short-Term Outcome Study; S.M.: advisory board, consultant (develop educational materials), Horizon Pharma; site PI, Kaleido Phase 2 Study; site PI Orphan Europe/Recordati NCGC Short-Term Outcome Study; site co-I Ultragenyx Clinical Trial; Translate Bio, consultant; advisory board, Arcturus; site PI for Prader–Willi syndrome clinical trials (active) Soleno, Levo, and Millendo; M.Y.: Translate Bio, consultant; Ultragenyx, consultant. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This report was prepared on behalf of the Urea Cycle Disorders Consortium, which, in addition to the authors, includes the following principal investigators: Nicholas Ah Mew, Mark L. Batshaw, Matthias R. Baumgartner, Gerard Berry, Peter Burgard, Lindsay Burrage, George A. Diaz, Gregory Enns, Renata C. Gallagher, Andrea Gropman, Cary O. Harding, Georg Hoffmann, Brendan Lee, J. Lawrence Merritt II, Sandesh C.S. Nagamani, Andreas Schulze, Tamar Stricker, Marshall L. Summar, Mendel Tuchman, Susan Waisbren, James D. Weisfeld-Adams, and Derek Wong.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Berry, S.A., Coughlin, C.R., McCandless, S. et al. Developing interactions with industry in rare diseases: lessons learned and continuing challenges. Genet Med 22, 219–226 (2020). https://doi.org/10.1038/s41436-019-0616-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0616-9
Keywords
This article is cited by
-
International comparison of pharmaceutical industry payment disclosures in the UK and Japan: implications for self-regulation, public regulation, and transparency
Globalization and Health (2023)
-
Together4RD position statement on collaboration between European reference networks and industry
Orphanet Journal of Rare Diseases (2023)
-
Genetic counseling in the context of Bangladesh: current scenario, challenges, and a framework for genetic service implementation
Orphanet Journal of Rare Diseases (2021)
-
Patient Preferences in Rare Diseases: A Qualitative Study in Neuromuscular Disorders to Inform a Quantitative Preference Study
The Patient - Patient-Centered Outcomes Research (2021)
-
Translational Genomics in Neurocritical Care: a Review
Neurotherapeutics (2020)
