
Abstract
Purpose
Krabbe disease (OMIM 245200) is an orphan neurometabolic disorder caused by a deficiency of the lysosomal enzyme galactocerebrosidase (GALC). Hard clinical endpoints and biomarker–phenotype correlations are useful for future clinical trials.
Methods
We performed a quantitative analysis of published cases (N = 248) with Krabbe disease, stratified by age at disease onset: early infantile (age 0–6 months), late infantile (age 7–36 months), juvenile/adolescent (age 37–180 months), and adult onset (>180 months). Main outcome measures were age of disease onset and survival. Cerebrospinal fluid (CSF) protein concentrations were explored as a potential predictor of survival. STROBE criteria were respected.
Results
Median age of onset was 4 months (early infantile), 14 months (late infantile), 48 months (juvenile), and 384 months (adult). Age of disease onset and therefore disease subtype determined survival rates. CSF protein concentrations predicted age at onset and survival rates in Krabbe disease. Patients with a CSF protein content ≤61.5 mg/dl survived significantly longer than patients with CSF protein values above this threshold.
Conclusion
We define the estimated survival in published Krabbe disease cases and demonstrate an association of CSF protein concentration with disease severity. These data inform patient care and clinical trials.
Similar content being viewed by others


INTRODUCTION
Krabbe disease (OMIM 245200) is an autosomal recessive neurodegenerative disease caused by deficiency of the lysosomal enzyme galactocerebrosidase (GALC, EC 3.2.1.46)1. The gene encoding galactocerebrosidase, GALC, is localized on chromosome 14q31.32. At present (9 October 2018), 237variants in GALC are included in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GALC). The prevalence is estimated to be between 1.0/100,000 and 1.0/250,0003,4.
In 1916, Knud Krabbe of Copenhagen described three patients from two families who showed spasticity with a progressive neurological deterioration in early infancy.5 Over time it became evident that some patients only develop symptoms late in their life, e.g., even in their fifties or later.6 Krabbe disease is therefore traditionally classified into different subtypes based on age at onset,7 e.g., early infantile, (late) infantile, late onset, and adult onset. However, definition of criteria for subtype classification and the nomenclature varies in the literature e.g., “late onset,” “late infantile,” “juvenile,” or “adult onset.”
Typical clinical features of “early infantile” or “early onset” Krabbe disease are feeding difficulties, hyperirritability, psychomotor regression, episodic fever of unknown origin, and convulsive seizures.7 Fundoscopic inspection can reveal macular cherry red spots.8 Later patients with early onset Krabbe disease reach the “burnout” stage with blindness, hypotonia, and decerebration without any voluntary movements. Both microcephaly and macrocephaly occur depending on the disease stage.7 The patients with onset other than early infantile, so-called late onset Krabbe disease, show various neurological symptoms such as ataxia, muscle weakness, blindness, spastic paraparesis, behavioral problems, and dementia.7
Furthermore, despite ongoing research efforts in recent decades, currently no drug is approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of Krabbe disease. The current understanding of the natural history of Krabbe disease is mainly based on three publications: the Hunter’s Hope Krabbe Family Database conducted a study based on questionnaires directed to families with afflicted individuals,9 and the World-Wide Krabbe Registry has collected natural history data, based on data from patients (self-referrals) and health-care providers (child neurologists or geneticists).10,11 The evidence of published case reports as a source for natural history data including survival time is not systematically explored. It is also not known whether the evidence embedded in published individual case reports is consistent with the natural history data based on questionnaires.
We therefore directed our research efforts at (1) quantitating hard clinical endpoints, such as onset of disease and survival; and (2) exploring potential biomarkers and investigating phenotype–biomarker associations in Krabbe disease, all based on the yet unexplored evidence in published case reports.
MATERIALS AND METHODS
This study was designed, executed, and reported in accordance with STROBE (STrengthening the Reporting of OBservational studies in Epidemiology) criteria.12
Literature review and definitions of variables
PubMed was searched using the keywords “Krabbe disease” and “galactocerebrosidase deficiency.” Identified publications were downloaded and manually sorted for reports containing relevant clinical, biochemical, and/or genetic data. Reports qualifying for further analysis were published between 1982 and 2017. In total, we identified 248 patients from 77 publications with sufficient information for analysis (Supplementary Figure 1). The articles were published in English, French, or Italian. Close of database was 30 December 2017. An inventory of the respective publications on Krabbe disease patients included in this analysis is provided in Supplementary Table 1. Publications were checked for duplicate reports.
The following variables were extracted: age at disease onset, mode of diagnosis (e.g., enzymatic, genetic), last reported age, information on whether the patient is alive or deceased, protein concentrations in cerebrospinal fluid (CSF), residual galactocerebrosidase (GALC) activity, publication year, and origin of patients. If the origin of the patient has not been explicitly stated in the report, the country of patient’s origin has been attributed to the country of the first author’s institutional affiliation in the respective case description.
If information regarding time was stated in semiquantitive terms, we took a conservative approach and defined the findings as follows: “stillborn” = day 0, “newborn” or “at birth” = day 1, “newborn period” = 1 month, “postmortem” = age at death.
Patients were considered alive at the time of the report if it was not explicitly stated that the patient was deceased. Patients with Krabbe disease are stratified into subtypes whereas the classification and terminology varies in the literature and subtypes overlap. To render our age-adjusted analyses unequivocal, we stratified reported patients into four age groups based on the available information about age at onset. The four groups were early infantile (age 0–6 months), late infantile (age 7–36 months), juvenile/adolescent (age 37–180 months), and adult onset (>180 months).
Statistical analysis
Techniques of descriptive statistics were applied as previously reported.13,14,15 Baseline patient demographics were summarized descriptively using patient counts and percentages of the total study population. Survival was assessed and defined as the time elapsed between patient birth and time of death. Patient data were censored at the time of last follow-up if the patient was still alive at last contact based on data gathered from each publication. Survival was estimated using the Kaplan–Meier method. The log-rank test was applied to compare potential differences between subgroups. We used unbiased recursive partitioning to determine cut-off values for the impact of CSF protein content on survival in subgroup analysis.16 To evaluate correlation between CSF protein concentrations and age of onset analysis of variance (ANOVA) was applied. We also analyzed the subtype of the patients based on the year of publication. For this analysis, year of publication was categorized into the following subgroups: 1981–1985, 1986–1990, 1991–1995, 1996–2000, 2001–2005, 2006–2010, 2011–2015, and 2016–2020. The world map was plotted using the R extension ggmap.17 Missing data were not imputed. Sensitivity analyses were not conducted. All analyses were performed using R (http://www.r-project.org). P values reported were two-sided, with P ≤ 0.05 considered statistically significant.
RESULTS
We identified 248 patients from 77 case descriptions or case series (published between 1982 and 2017) for further statistical analysis. The characteristics of the study cohort are depicted in Table 1. The origin of afflicted individuals is illustrated in Fig. 1, which indicates a panethnic distribution pattern for Krabbe disease. Due to missing data in different subsets, patients had to be excluded for specific subsequent data analyses. Sample sizes (N) are always indicated for the corresponding analyses.

Distribution pattern of patients with Krabbe disease. Gray scale indicates the number of identified affected individuals per country. Light gray: 1 to 10 patients, medium gray: 11 to 40 patients, dark gray: more than 40 patients.
Confirmation of diagnosis of Krabbe disease
The diagnosis of Krabbe disease was confirmed by either enzymatic analysis of galactocerebrosidase activity in 111 patients (44.8%) or by variant analysis of the GALC gene in 1 patient (0.4%) alone. In 130 patients (52.4%), a combination of enzymatic and genetic analysis was performed. In 6 patients (2.4%), neither enzymatic nor genetic analyses were explicitly reported in the publications. In those cases, diagnosis of Krabbe disease was established by characteristic histological findings (e.g., globoid cells), affectedness of a sibling with either enzymatically or genetically confirmed Krabbe disease, or morphological alterations on cranial magnetic resonance imaging (cMRI) scans characteristic of Krabbe disease.18,19,20,21
Stratification into subgroups based on age of disease onset
Because the patient populations in the original descriptions—i.e., early infantile, late infantile, juvenile, and adult—partially overlap, we stratified the population into four groups based on age at onset for subsequent analysis: 0–6 months (early infantile), 7–36 months (late infantile), 37–180 months (juvenile/adolescent), and older than 180 months (adult onset) (Supplementary Table 2). After recategorization, 98 (39.5%) patients belonged to the early infantile, 57 (23%) to the late infantile, 46 (18.5%) to the juvenile/adolescent, and 42 (16.9%) to the adult onset group. For 5 (2.1%) patients, the age of disease onset was not reported (Table 1). A stratified distribution pattern of Krabbe disease patients by subgroup and continent is depicted in Supplementary Table 3.
Age of disease onset
We quantitatively analyzed the age of disease onset in stratified subgroups. Median age of onset was 4 months, interquartile range (IQR) from 3 to 5 months for the early infantile group (N = 98); 14 months, IQR from 10 to 24 months for the late infantile group (N = 57); 48 months, IQR from 48 to 72 months for the juvenile group (N = 46); and 384 months, IQR from 165.9 to 516 months for the adult onset group (N = 42).
Survival estimations
In the overall population, 184 patients (74.2%) patients were alive, whereas 64 patients (25.8%) were deceased at the time of publication (Table 1). Estimated survival analysis using the Kaplan–Meier method showed that 54.1% patients with Krabbe disease were still alive at the age of 19 years for the whole study population (Supplementary Figure 2). As hypothesized, age of disease onset and therefore Krabbe disease subtype determined survival rates of afflicted individuals; the median survival for the early infantile group was 1.5 years (N = 67) and for the late infantile group 9.5 years (N = 38) (Fig. 2a). In the juvenile group 80% of patients were alive at the age of 16 years (N = 35), whereas in the adult onset group 87.9% of patients were still alive at the age of 19 years (N = 33) (Fig. 2a). Survival was statistically significantly shorter in early infantile compared with late infantile, juvenile, and adolescent onset groups (i.e., early infantile versus late infantile, late infantile versus juvenile, juvenile versus adult; p < 0.001, log-rank test).

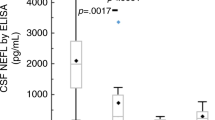
Estimated survival distributions and age of disease onset for patients with Krabbe disease. (a) Estimated survival distribution for patients with Krabbe disease per subtype (N = 173). Censored individuals are marked with a “+.” (b) Estimated survival distribution for Krabbe disease patients with a cerebrospinal fluid (CSF) protein concentration above 61.5 mg/dl (N = 34, black line) and below or equal to 61.5 mg/dl (N = 14, gray line). Censored individuals are marked with a “+.” Log-rank test, p = 0.002. (c) Age of disease onset/Krabbe disease subtype subject to CSF protein content (mg/dl). Data were available for N = 28 patients in the early infantile, N = 12 in both late infantile and juvenile groups and N = 7 in the adult onset group. Black lines indicate the median per group, single dots individual extreme values.
CSF protein concentration
Protein levels in the CSF were determined in 24% of patients (59/248). Intriguingly, patients with a protein concentration below or equal to 61.5 mg/dl in CSF survived significantly longer as opposed to patients whose CSF protein levels were above this threshold (p = 0.002, log-rank test; N = 48; threshold determined by unbiased recursive partitioning) (Fig. 2b). Moreover, CSF protein concentration inversely correlated with the age of disease onset, e.g., CSF protein content was significantly higher in the early infantile group when compared with the late infantile, juvenile, and adult onset groups (p < 0.01, ANOVA with Tukey–Kramer test, N = 59) (Fig. 2c).
Residual GALC activity
Because in other lysosomal storage diseases, residual enzymatic activities correlate with and predict survival rates and/or age of disease onset,13,15 we investigated whether this also holds true for residual GALC activity in individuals with Krabbe disease. In our cohort, residual GALC activity in all investigated specimens (i.e., serum, dried blood spots, leukocytes, fibroblasts, CSF) correlated neither with age of disease onset nor survival.
Descriptive statistics of CSF protein concentrations and residual GALC activity in different specimens are summarized in Supplementary Table 4.
Distribution of Krabbe disease subtypes by year of publication
We analyzed patient distribution of Krabbe disease subtypes by year of publication according to defined subgroups starting in 1981 to the current date in 5-year steps. The number of overall reported patients in 2011–2015 was dramatically increased (N = 90) compared with the number of patients reported in 1981–1985 (N = 6) as indicated by the width of the corresponding columns in the spinogram (Fig. 3). Furthermore, the distribution pattern of Krabbe disease subtypes substantially changed over the last three decades with a higher incidence of adult onset cases after the year 2010; in the subgroup 1981–1985, 83.3% (N = 5) of the reported patients belonged to the early infantile and 16.7% (N = 1) to the juvenile/adolescent Krabbe disease subtype, whereas in 2011–2015 reported patient distribution pattern evolved with 37.8% (N = 34) in the early infantile, 25.6% (N = 23) in the late infantile, 13.3% (N = 12) in the juvenile, and 23.3% (N = 21) in the adult onset group (Fig. 3).

Spinogram: distribution of Krabbe disease subtypes by year of publication. The spinogram illustrates the proportion of patients reported attributed to disease subtype (early infantile, late infantile, juvenile, and adult onset) per 5-year time intervals. “(“ indicates that the corresponding year has been excluded; “]” indicates that the corresponding year has been included into the respective subset. The width of each column depicts the number of case descriptions reported within the respective time intervals.
DISCUSSION
We describe the natural history of Krabbe disease based on a large sample of 248 patients from 77 case reports in the literature, published between 1982 and 2017. By size, this adds substantial evidence to the known natural history information on Krabbe disease: the reported data provided by the Hunter’s Hope family database were based on 334 returned family questionnaires of variable completeness,9 and the summary data published from the World-Wide Krabbe Registry contained information from 180 patients.10,11,22 The patient population in our study appears to be biased to later disease onset: only 39.5% of patients in the present study had early infantile disease onset (before 6 months), whereas 71% of the Hunter’s Hope family database, and 62% of the World-Wide Krabbe Registry patients had disease onset at this age.9,10 In our study, 23% had disease onset between 7 and 36 months, 18.5% between 37 and 180 months, and 16.9% after 180 months. In the previously reported questionnaire-based study, 19% of patients had disease onset between 7 and 12 months and 10% between 13 months and 5.5 years.9 In the World-Wide Krabbe Registry, 10% of patients manifested Krabbe disease at 7–12 months, 22% between 13 months and 10 years, and 5% were adolescent/adult.11
In this study, the median age at onset of disease in the early infantile group was 4 months. This is consistent with the age of disease onset in the early infantile group reported in the Krabbe registry, which was between 2 and 5 months.10 Estimated survival analysis of the whole study sample using the Kaplan–Meier method showed that 54.1% of Krabbe disease patients were still alive at the age of 19 years. Importantly, age at onset and therefore disease subtype was a statistically significant predictor of survival (p < 0.01).
It has been reported that CSF protein concentrations are high in patients with “infantile” and “late infantile” Krabbe disease, and normal or only moderately elevated in juvenile or adult patients. However, this statement is based on seven patients from four publications.23,24,25,26 Our study, which analyzed strictly categorized subgroups based on age of onset, showed that an earlier onset of disease was strongly associated with a higher CSF protein concentration (p < 0.01). Moreover, protein content in CSF clearly predicted survival rates: patients with a protein content below or equal to 61.5 mg/dl survived significantly longer than patients with CSF protein concentrations above this threshold.
CSF protein concentration qualifies as a biomarker as it somewhat reflects the activity of the disease process; in the case of Krabbe disease, probably a disturbed blood–brain barrier. CSF protein may be considered a diagnostic biomarker in the appropriate clinical context of Krabbe disease. To qualify as a true surrogate marker, it is necessary that it captures the severity of disease, which may be the documented association with age at onset and survival in this study. In addition, it is necessary that the net effect of treatment on the true clinical outcome is demonstrated in therapeutic trials.27,28 Possible confounders of CSF elevations may be unspecific including central nervous system (CNS) infections, demyelinating diseases, and malignancies, which may limit its usefulness as a biomarker for Krabbe disease. Recently, Escolar et al. proposed psychosine concentrations in dried blood spots as a biomarker, which reflected disease activity and can be used for diagnostic and pharmacodynamic purposes.29 Psychosine, also known as galactosylsphingosine, is a substrate of the GALC enzyme and is considered to be toxic for glial and neuronal cells.30
Residual GALC enzyme activities in all investigated specimens did not have any predictive impact on survival rates or on the phenotypic presentation assessed by the variable age at disease onset. In line with our findings, Tappino et al. did not observe a relationship between GALC activity and age at onset.31 In contrast, Jalal et al. found that higher residual enzyme activities, i.e., above 0.10 nmol/h/mg protein, were not observed in patients with the early infantile variant of Krabbe disease.32
Experimental therapeutic approaches are focused on hematopoietic stem cells whereas the therapeutic “window of opportunity” has narrow time margins.33 New York State started screening all newborns for Krabbe disease in 2006. Of 2 million babies, 5 were identified to have early infantile Krabbe disease; 1 died untreated, 2 died from hematopoietic stem cell transplantation (HSCT)-related complications, and 2 children having received HSCT survived with moderate to severe developmental delay.34 Forty-six children were identified as “at risk” for late onset Krabbe disease, which raises ethical issues.34 Clinicaltrials.gov currently lists nine completed studies for Krabbe disease (as of 11 July 2018, Supplementary Table 5). The publicly available results of the interventional studies add little evidence for the therapeutic management of the disease. Currently, there is no FDA or EMA approved therapy for the disease. Two investigational compounds were granted orphan drug designation by the FDA.35 Recombinant human galactocerebrosidase (rhGALC), an enzyme replacement therapy (ERT), received orphan drug designation in 2011 and ibudilast in 2015. Both compounds are not approved for treatment of Krabbe disease by the FDA (as of 11 July 2018). Recombinant human galactocerebrosidase was assigned orphan drug designation by the EMA in 201136.
Eighteen studies are currently open to enrollment (as of 11 July 2018, Supplementary Table 6). Preclinical data on positive effects of ERT on the twitcher mouse model were published in 200537; however, a prospective natural history study (NCT01093105) was withdrawn in 2010, and to date, there is currently no open clinical trial for rhGALC listed on clinicaltrials.gov. Ibudilast is a small molecule that inhibits the phosphodiesterase and macrophage migration inhibition factor, potentially resulting in anti-inflammatory and neuroprotective effects, approved for asthma and poststroke symptoms in Japan and other parts of Asia.38 Ibudilast was granted orphan designation for the treatment of Krabbe disease by the FDA in the United States. In the European Union, the compound has received orphan drug status for amyotrophic lateral sclerosis (ALS), but not yet for Krabbe disease. Currently, 13 clinical studies with ibudilast are listed on clinicaltrials.gov for various potential neurological and/or neurodegenerative indications (including ALS, migraine, headache, substance dependence, and multiple sclerosis, as of 11 July 2018). The geographical distribution pattern of reported patients suggests a panethnic pattern across all continents including developed and developing countries. Data on patient distribution might be of special interest for the planning of future prospective natural history and potential therapeutic studies with regard to patient recruitment.
Limitations
The present method, the definition of the natural history of an ultrarare condition through systematic analysis of published natural history cases, has successfully been applied in other conditions (mucopolysaccharidosis type VII,13 Farber disease,15 and free sialic acid storage disease14) where comprehensive prospective natural history studies would either be unfeasible or take many years to complete in a worldwide multicenter setting. This study’s approach has several very important limitations that have to be taken into account to place these data into the overall global context. The survival estimations are, at least in part, historical as they are based on studies published between 1982 and 2017. It is impossible to quantitate the bias due to change of supportive care. This bias could theoretically focus on two directions: either that supportive care has improved, which would have extended survival, or that a more palliative approach, centered on quality of life through less “interventional aggressiveness” rather than on prolongation of life at any cost, would have formally decreased survival in the natural history over time. As case reports often focus on a particular aspect of the disease, the case descriptions are not uniform and often lack details of interest. This renders the extraction of softer, but nevertheless important, endpoints such as cognitive development and physical features very imprecise.13,39 This limitation—ascertainment bias—is true for the questionnaire-based study as well as for the registry when either retrospective data are captured or a particular variable of interest is not prospectively assessed by protocol. Because of this substantial assessment bias we did not perform an in-depth analysis of softer endpoints. Recently, a prospective natural history study was published, describing important characteristics on soft clinical endpoints such as disease progression with regard to anthropometric data, changes in muscle tone and reflexes, as well as visional and hearing impairment in a cohort of 35 patients with Krabbe disease with onset between 6 months and 3 years of life.40 There has been some disagreement in the literature about the precise classification of Krabbe disease subtypes, especially the late onset forms. A clear-cut separation appears difficult, which is illustrated by the fact that the terminology varies and overlaps in part. A precise overview about the nomenclature used is presented in Supplementary Table 2.
While a prospective natural history study approach reliably defines softer clinical endpoints and biomarker–phenotype associations due to the implementation of a standardized protocol, it is associated with a high logistic burden and requires the availability of necessary resources in terms of time and personnel, which may not be feasible for every single one of the plethora of orphan diseases.14 Natural history case reports, on the other hand, may be subject to publication bias, i.e., researchers may tend to publish older, more atypically presenting patients, as journals may find nonconfirmatory information more “novel” and more interesting for their readers and, therefore, more acceptable for publication. Specifically, the year of publication introduced a bias into the distribution pattern of Krabbe subtypes. While it had become widely accepted in the early literature that early onset Krabbe disease was severe, the incentive to publish early onset case reports decreased over time. In the last 20 years, adult onset Krabbe disease was a relatively novel finding and publication pressure was then redirected to older patients. Missing data may be an issue for quantitative retrospective natural history modeling as outlined previously.14 The data completeness for hard endpoint variables such as survival was above 80% in this analysis, whereas the data availability for the explored softer, biochemical variables was lower, but there were nevertheless N = 59 patients available for analysis, which is a substantial sample size in the context of a rare disease (Supplementary Table 7).
Another important limitation is the fact that laboratory data were not obtained with standardized measurements run in a centralized laboratory, but were pooled from the information extracted in the published case reports. This introduces some statistical noise. It cannot be formally excluded that data from at least some of the patients in our study were also part of summary reports provided in the questionnaire-based study or the World-Wide Krabbe Registry. In N = 6/248 patients, an enzymatic and/or genetic confirmation of Krabbe disease was not explicitly stated in the peer-reviewed publications.18,19,20,21 We consider the diagnosis of Krabbe disease as correct, because the findings in these patients (e.g., clinical, morphological, histochemical) are consistent with Krabbe disease. The present study supports and corroborates the present evidence on the natural history of Krabbe disease based on questionnaires and the disease registry.9,10,11 Given these limitations, our approach was the most rapid and feasible method to define important outcome measures that will inform future clinical studies in Krabbe disease.
Conclusions
In this report, we provide natural history data including geographical localization of 248 published patients with Krabbe disease. We define the estimated survival and demonstrate an association of CSF protein concentration with disease severity, which may be an interesting biomarker for future clinical trials.
References
Suzuki K, Suzuki Y. Globoid cell leucodystrophy (Krabbe’s disease): deficiency of galactocerebroside beta-galactosidase. Proc Natl Acad Sci U S A. 1970;66:302–309.
Cannizzaro LA, Chen YQ, Rafi MA, Wenger DA. Regional mapping of the human galactocerebrosidase gene (GALC) to 14q31 by in situ hybridization. Cytogenet Cell Genet. 1994;66:244–245.
Orphanet Report Series. Prevalence of rare diseases: bibliographic data. June 2018, no. 1. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Accessed 11 July 2018.
Barczykowski AL, Foss AH, Duffner PK, Yan L, Carter RL. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomal storage diseases. Am J Med Genet A. 2012;158A:2835–2842.
Krabbe K. A new familial, infantile form of diffuse brain. sclerosis. Brain. 1916;39:74–114.
Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci. 1991;13:232–239.
Wenger DA, Escolar ML, Luzi P, Rafi MA Chapter 147 - Krabbe disease (Globoid Cell Leukodystrophy) In: Valle D, (ed.) The Online Metabolic and Molecular Bases of Inherited Disease. 2018. https://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62644214. Accessed 11 July 2018.
Naidu S, Hofmann KJ, Moser HW, Maumenee IH, Wenger DA. Galactosylceramide-beta-galactosidase deficiency in association with cherry red spot. Neuropediatrics. 1988;19:46–48.
Duffner PK, Jalal K, Carter RL. The Hunter’s Hope Krabbe family database. Pediatr Neurol. 2009;40:13–18.
Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM, Carter RL. Early infantile Krabbe disease: results of the World-Wide Krabbe Registry. Pediatr Neurol. 2011;45:141–148.
Duffner PK, Barczykowski A, Kay DM, et al. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatr Neurol. 2012;46:298–306.
von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370:1453–1457.
Zielonka M, Garbade SF, Kolker S, Hoffmann GF, Ries M. Quantitative clinical characteristics of 53 patients with MPS VII: a cross-sectional analysis. Genet Med. 2017;19:983–988.
Zielonka M, Garbade SF, Kolker S, Hoffman GF, Ries M. A cross-sectional quantitative analysis of the natural history of free sialic acid storage disease—an ultra-orphan multisystemic lysosomal storage disorder. Genet Med. 2019;21:347–352.
Zielonka M, Garbade SF, Kolker S, Hoffmann GF, Ries M. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genet Med. 2018;20:524–530.
Hothorn T, Hornik K, Zeileis A. Unbiased recursive partitioning: a conditional inference framework. J Comput Graph Stat. 2006;15:651–674.
Kahle D, Wickham H. ggmap: spatial visualization with ggplot2. R J. 2013;5:144–161.
Fiumara A, Barone R, Arena A, et al. Krabbe leukodystrophy in a selected population with high rate of late onset forms: longer survival linked to c.121G>A (p.Gly41Ser) mutation. Clin Genet. 2011;80:452–458.
Hittmair K, Wimberger D, Wiesbauer P, Zehetmayer M, Budka H. Early infantile form of Krabbe disease with optic hypertrophy: serial MR examinations and autopsy correlation. AJNR Am J Neuroradiol. 1994;15:1454–1458.
Jones BV, Barron TF, Towfighi J. Optic nerve enlargement in Krabbe’s disease. AJNR Am J Neuroradiol. 1999;20:1228–1231.
Shah S, Freeman E, Wolf V, Murthy S, Lotze T. Intracranial optic nerve enlargement in infantile Krabbe disease. Neurology. 2012;78:e126.
Langan TJ, Barcykowski AL, Dare J, Pannullo EC, Muscarella L, Carter RL. Evidence for improved survival in postsymptomatic stem cell-transplanted patients with Krabbe’s disease. J Neurosci Res. 2016;94:1189–1194.
Epstein MA, Zimmerman RA, Rorke LB, Sladky JT. Late-onset globoid cell leukodystrophy mimicking an infiltrating glioma. Pediatr Radiol. 1991;21:131–132.
Goebel HH, Harzer K, Ernst JP, Bohl J, Klein H. Late-onset globoid cell leukodystrophy: unusual ultrastructural pathology and subtotal beta-galactocerebrosidase deficiency. J Child Neurol. 1990;5:299–307.
Grewal RP, Petronas N, Barton NW. Late onset globoid cell leukodystrophy. J Neurol Neurosurg Psychiatry. 1991;54:1011–1012.
Phelps M, Aicardi J, Vanier MT. Late onset Krabbe’s leukodystrophy: a report of four cases. J Neurol Neurosurg Psychiatry. 1991;54:293–296.
Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx. 2004;1:189–195.
Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–440.
Escolar ML, Kiely BT, Shawgo E, et al. Psychosine, a marker of Krabbe phenotype and treatment effect. Mol Genet Metab. 2017;121:271–278.
Spassieva S, Bieberich E. Lysosphingolipids and sphingolipidoses: psychosine in Krabbe’s disease. J Neurosci Res. 2016;94:974–981.
Tappino B, Biancheri R, Mort M, et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat. 2010;31:E1894–1914.
Jalal K, Carter R, Yan L, Barczykowski A, Duffner PK. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatr Neurol. 2012;47:324–329.
Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069–2081.
Wasserstein MP, Andriola M, Arnold G, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med. 2016;18:1235–1243.
US Food and Drug Administration. Search orphan drug designations and approvals, 2017. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/. Accessed 11 July 2018.
European Medicines Agency. Public summary of opinion on orphan designation. Recombinant human galactocerebrosidase for the treatment of globoid cell leukodystrophy (Krabbe disease). 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2011/10/WC500116522.pdf. Accessed 11 July 2018.
Lee WC, Courtenay A, Troendle FJ, et al. Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 2005;19:1549–1551.
Fox RJ, Coffey CS, Cudkowicz ME, et al. Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp Clin Trials. 2016;50:166–177.
Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986–1996.
Bascou N, DeRenzo A, Poe MD, Escolar ML. A prospective natural history study of Krabbe disease in a patient cohort with onset between 6 months and 3 years of life. Orphanet J Rare Dis. 2018;13:126.
Acknowledgements
No funding was secured for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
M.R. received consultancy fees or research grants from Alexion, GSK, Oxyrane, and Shire. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Komatsuzaki, S., Zielonka, M., Mountford, W.K. et al. Clinical characteristics of 248 patients with Krabbe disease: quantitative natural history modeling based on published cases. Genet Med 21, 2208–2215 (2019). https://doi.org/10.1038/s41436-019-0480-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0480-7
Keywords
This article is cited by
-
A novel mutation in the GALC gene causes Krabbe disease accompanied with extensive Mongolian spots in a consanguineous family
Neurological Sciences (2023)
-
A novel compound heterozygous mutation in GALC associated with adult-onset Krabbe disease: case report and literature review
neurogenetics (2022)
-
Two Cases of Female Chinese Adult-Onset Krabbe Disease with One Novel Mutation and a Review of Literature
Journal of Molecular Neuroscience (2021)
