
Abstract
Purpose
Haploinsufficiency of USP7, located at chromosome 16p13.2, has recently been reported in seven individuals with neurodevelopmental phenotypes, including developmental delay/intellectual disability (DD/ID), autism spectrum disorder (ASD), seizures, and hypogonadism. Further, USP7 was identified to critically incorporate into the MAGEL2-USP7-TRIM27 (MUST), such that pathogenic variants in USP7 lead to altered endosomal F-actin polymerization and dysregulated protein recycling.
Methods
We report 16 newly identified individuals with heterozygous USP7 variants, identified by genome or exome sequencing or by chromosome microarray analysis. Clinical features were evaluated by review of medical records. Additional clinical information was obtained on the seven previously reported individuals to fully elucidate the phenotypic expression associated with USP7 haploinsufficiency.
Results
The clinical manifestations of these 23 individuals suggest a syndrome characterized by DD/ID, hypotonia, eye anomalies,feeding difficulties, GERD, behavioral anomalies, and ASD, and more specific phenotypes of speech delays including a nonverbal phenotype and abnormal brain magnetic resonance image findings including white matter changes based on neuroradiologic examination.
Conclusion
The consistency of clinical features among all individuals presented regardless of de novo USP7 variant type supports haploinsufficiency as a mechanism for pathogenesis and refines the clinical impact faced by affected individuals and caregivers.
Similar content being viewed by others


INTRODUCTION
De novo heterozygous loss-of-function pathogenic variants in the gene ubiquitin-specific protease 7 (USP7, also called herpes virus-associated ubiquitin-specific protease, HAUSP), were recently identified in seven individuals with a novel neurodevelopmental disorder, manifesting intellectual disability, autism spectrum disorder (ASD), and seizures.1 Six of these individuals had genomic heterozygous deletions involving USP7. One more individual with a nonsense variant of USP7 had a similar phenotype, indicating a haploinsufficiency disease model.
Located at chromosome 16p13.2, USP7 encodes a deubiquitinating proteolytic enzyme that can cleave multiple ubiquitin chain linkages.2,3,4 Previously, USP7 was shown to regulate ubiquitination of proteins including the MDM2-p53 pathway, important for DNA repair, transcription, and cancer.2,3
USP7 was reported to function in the endosomal protein recycling pathway via its incorporation into the MAGEL2-USP7-TRIM27 (MUST) complex.1,5 USP7 was found to promote ubiquitination of the actin nucleation promoting factor WASH by preventing TRIM27 autoubiquitination and subsequent degradation, while simultaneously limiting WASH ubiquitination through direct deubiquitination of WASH.1 USP7’s dual function allows the fine-tuning of WASH activity, maintaining an appropriate level of endosomal F-actin and a homeostatic balance for retromer-dependent protein recycling.1,5,6
Here we report 16 newly identified individuals with de novo pathogenic variants in USP7. Among these, we present two individuals with a deletion of USP7, three individuals with nonsense variants, three with splice-site variants, and eight with missense variants. We also provide expanded clinical information on the seven previously reported individuals.
MATERIALS AND METHODS
Individuals with USP7 pathogenic variants were enrolled based on genotype. Identification of patients 1–6 and 9 was described previously (patients 1–5 have retained the same labels; patient 6 was previously labeled subject 7; patient 9 was previously labeled subject 6) (ref. 1). The 16 newly identified probands were identified by clinical genome sequencing or exome sequencing (ES) or chromosomal microarray (CMA) (coordinates based on build hg19). Upon identification of a probable pathogenic variant or deletion of USP7, genetic counselors/physicians from the respective laboratories and institutions contacted the referring providers, informing them of a research interest in C.P.S.’s laboratory. Individuals were either placed in contact with M.D.F. and C.P.S. upon molecular genetic confirmation of a pathogenic variant of USP7, referred to them via the Foundation for USP7-Related Diseases contact pages (www.USP7.org), or identified and enrolled through GeneMatcher. One individual was identified via a personal blog post describing primary clinical manifestations and molecular genetic testing results.
An initial phenotypic assessment was obtained of all newly identified and molecularly confirmed individuals harboring pathogenic variants/deletions of USP7 (patients 1–23 presented in Tables S1–S4). Patients 18 and 20 harbor additional variants in TMEM106B and SLC2A1, respectively, but were nonetheless enrolled. Likewise, patient 19 also has a de novo heterozygous 102.5-kb mosaic loss of uncertain significance at 10q21.1 but was also enrolled. Upon consent to share clinical information, all were contacted for a detailed review of medical and family history, developmental milestones, and physical features. Medical records were reviewed as comprehensively as possible. Written informed consent was obtained for use of medical history, genetic testing report, and photograph (if applicable), as approved by the Baylor College of Medicine Institutional Review Board.
Statistical analysis of corpus callosum biometry (further discussed below) was performed by calculating age-based domain-specific Z-scores for all cases using available normative data. To evaluate differences between children with USP7 variants and normative data, a one-sample Z-test was used to determine if the mean Z-score for the cases differed from the normative mean of zero. Differences were considered statistically significant if P < 0.05. All statistical analyses were conducted using Stata version 15 (College Station, TX).
RESULTS
Here we present 16 newly identified individuals with a novel neurodevelopmental disorder, all of whom have molecularly confirmed heterozygous de novo variants in USP7. We also provide expanded clinical information on the seven previously reported individuals.1 All individuals’ behavioral and physical characteristics are summarized in Table 1; subjects’ genomic information is provided in Table 2. Each individual’s clinical phenotype is provided in Tables S1, S2, S3, and S4.
Behavioral characteristics
All individuals with USP7 variants have prominent speech delays. Moreover, 18% of individuals are currently nonverbal (patient 4, 10 years old; patient 13, 8 years old; patient 15, 4 years old; and patient 17, 20 years old). Regarding motor milestones, on average, affected children sat independently at 14 months (range: 4–36 months) and walked at 35 months (range: 17–156 months, patient 15 not walking).
Of those with formal evaluation, intellectual disability severity ranged from borderline intellectual functioning (with one patient reporting a standardized intelligence quotient [IQ] score in the 80s) to mild–moderate intellectual disability (with one patient reporting a standardized IQ score of 52, and one reporting a standardized nonverbal IQ test score of 74). An additional six patients had not been formally evaluated or did not have results available but were noted to require special education.
Of the 17 individuals with available data, five were formally diagnosed with ASD, with an additional four described to manifest autistic features.
Four of seven individuals were previously identified to have aggressive behaviors by their referring physician.1 Further evaluation of previously identified and new patients identified an expanded behavioral profile to include impulsivity, compulsivity, stubbornness, manipulative behaviors, and a propensity for temper tantrums and aggressive behaviors in 12 of 21 individuals. Additionally, attention-deficit hyperactivity disorder (ADHD) was present in 6 (4 diagnosed, 2 with signs) of 16 individuals.
Physical characteristics
While 18 of 20 individuals have facial dysmorphisms, including deep-set eyes and a prominent nasal septum extending below the alae nasi, these features are variable and do not currently represent a recognizable dysmorphic condition (Fig. 1). Seven of 16 individuals manifest hypogonadism (5 males with cryptorchidism and/or micropenis, and 2 females with delayed development of secondary sex characteristics). Twelve of 22 have a history of feeding problems, and 8 of 16 report gastroesophageal reflux disease (GERD). Ten of 22 report seizures. Upon radiologic examination, 11 of 15 individuals manifested abnormal brain magnetic resonance imaging (MRI) results, further discussed below.

Facial appearance of patients with haploinsufficiency of USP7. While most individuals have facial dysmorphisms, including deep-set eyes and a prominent nasal septum, extending below the alae nasi, the dysmorphic features present across a varied spectrum and do not represent a recognizable dysmorphic condition at this time. (a) Patient 3. (b) Patient 7. (c) Patient 8. (d) Patient 10. (e) Patient 11. (f) Patient 13. (g) Patient 14. (h) Patient 15. (i) Patient 16. (j) Patient 17. (k) Patient 18. (l) Patient 19. (m) Patient 21. (n) Patient 22.
Molecular characteristics
All 23 individuals reported here have molecularly confirmed de novo heterozygous deletions/variants involving USP7 (Table 2). Eight individuals, six previously described,1 have deletions of chromosome 16 including all or part of USP7, detected by CMA or genome sequencing. Four individuals, one previously reported,1 have nonsense variants in USP7, identified via ES. Eight newly identified individuals have missense variants in USP7, and three newly identified individuals have splice-site variants in USP7, all identified via ES.
Deletion of USP7
Eight individuals harbor whole or partial deletions of USP7 (Fig. 2a, Table 2). Patients 1–6 were reported previously, and patients 7 and 8 are newly identified individuals. All eight manifest DD/ID and speech delays; one is nonverbal. Strikingly, seven of eight manifest a characteristic behavioral profile, and seven of seven individuals (five previously reported and two newly identified) manifest ASD or autistic features. Though ASD was previously endorsed for patients 2, 4, and 5, upon further chart review they were found only to exhibit autistic features (Table 1). Of note, patient 1 was previously reported not to have aggressive behavior, but after further review was found to exhibit the characteristic behavioral phenotype. Additionally, patient 3 was previously reported to demonstrate aggressive behavior but upon additional review was found only to show some signs of the behavioral phenotype.1

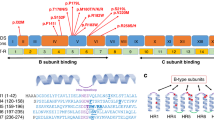
USP7 deletions and point variants. (a) USP7 whole and partial gene deletions in eight individuals, six of which were previously reported, identified via chromosomal microarray (CMA) or genome sequencing. Asterisks denote previously described individuals; labels used in the previous report are indicated by numbers in parentheses.1 Newly identified individuals are formatted in bold. Blue and green bars show the position of the deletions. Genomic coordinates are based on the Human Genome Assembly hg19. (b) Nonsense variants, frameshifting indels, and missense variants in USP7. Two individuals, one of whom was previously reported, have nonsense variants in USP7. Two newly identified individuals have frameshifting indels of USP7. Eight newly identified individuals have missense variants in USP7. Bolded variants indicate newly identified individuals; the nonbolded variant indicates a previously reported case. All variants based on NM_003470.2. In addition, three newly identified individuals with splice-site variants in USP7 are reported in the manuscript, which are not represented visually in this figure.
Six of eight individuals manifest eye anomalies, specifically myopia, esotropia, and strabismus, and four of six show dysmorphic facial features. Five of seven manifest hypogonadism; specifically, four males show cryptorchidism and/or micropenis, and one female shows delayed development of secondary sex characteristics and no regular menses.
Though only one of six individuals was noted to have neonatal hypotonia, four of seven manifested hypotonia at the time of exam (though patient 4 had low-normal tone). Subject 3 was previously reported not to manifest hypotonia; however, further examination of her chart and physical history did reflect hypotonia.1 Interestingly, patient 8 was reported to manifest hypertonia.
Three of four individuals showed gross neurologic aberrations/alterations based upon MRI, characterized by a paucity of white matter and atypical corpus callosum, and four of eight manifested seizures.
Nonsense variants in USP7
Four individuals have nonsense variants in USP7 (Fig. 2b, Table 2). Patient 9 was previously reported as subject 6 (ref. 1); all others are newly identified. Patients 9 and 10 have a nonsense pathogenic variant, patient 11 has a frameshift insertion, and patient 12 has a frameshift deletion. All manifest DD/ID and speech delays (though all are verbal), and one of three manifests the characteristic behavioral profile; however, none shows ASD or ADHD.
All four individuals had hypotonia neonatally and at the time of exam, eye anomalies, and dysmorphic facial features. Two of the four manifest feeding difficulties requiring assistance; in addition, patient 9 had decreased fetal movement and infantile lethargy, difficulty gaining weight, chronic constipation, and GERD. However, the true prevalence of these characteristics is unclear, as information for the above difficulties was unavailable for several of the three other patients.
Neuroradiologic examination showed abnormalities in one of three patients; patient 9 showed decreases in white matter, patients 10 and 12 showed no MRI anomalies, and patient 11 has not had MRI performed. Two of four individuals reported seizures.
Missense variants in USP7
Of the 23 total individuals, 8 (2 males, 6 females) have missense variants in USP7 (Fig. 2b, Table 2). All eight manifest speech delays, with three being nonverbal. Seven of eight manifest DD/ID. Two of seven manifest the characteristic behavioral profile. One of three assessable individuals manifests ASD, and one additional individual shows autistic features. One of four manifests ADHD.
Five of seven manifested hypotonia at the time of exam, while four of seven manifested neonatal hypotonia. Seven of seven manifest characteristic facial features, and four of the eight manifest eye anomalies, namely strabismus, nystagmus, and aniscoria. Four of the eight were reported to have feeding difficulties requiring assistive techniques, with five of seven individuals having difficulty gaining weight, four of six reporting GERD, and two of five manifesting chronic diarrhea. Four of four individuals have an abnormal gait. Two of four manifest features of hypogonadism, with one male manifesting cryptorchidism and one female manifesting labial hypoplasia.
Neuroradiologic examination identified MRI anomalies in seven of seven individuals; all showed decreased white matter, and patient 19 also manifested hydrocephalus ex vacuo and ventriculomegaly. Three of seven individuals have a history of seizures.
Splice-site variants in USP7
Three newly identified individuals (all male) have splice-site variants in USP7 (Fig. 2b, Table 2). Individuals 22 and 23 are siblings. All manifest DD/ID and speech delays, though all are verbal (Table 1). Two of the three show the characteristic behavioral profile as well as ADHD features, though none manifests ASD.
All show dysmorphic facial features, one of three manifests eye anomalies (strabismus), and one of three manifested hypotonia at the time of exam but none reported hypotonia neonatally. Two of three manifest feeding problems, though none reported associated problems such as neonatal poor suck, GERD, chronic constipation, chronic diarrhea, or difficulty gaining weight; in contrast, all three individuals show excessive weight gain. Two of three also reported sleep disturbances and asthma.
One individual manifests kyphosis and the two others manifest scoliosis. None shows hypogonadism or cryptorchidism.
One individual (patient 22) of the three reported suspected seizures. Only one patient (23) had MR imaging performed, which was reported to be normal.
Neuroradiologic examination
Brain MRI analysis and/or chart review identified neuroradiologic anomalies in 11 of 15 individuals, with primary features of decreased white matter and thinning of corpora callosa. Specifically, chart review of four individuals (2, 5, 9, and 19) noted neuroradiologic anomalies via MRI to include decreased white matter; in addition, periventricular leukomalacia was noted in patient 5. Chart review of MRI analysis for patients 4, 10, 12, and 23 noted no abnormalities. The imaging for these eight individuals was not available for secondary analysis. MR imaging and results for seven more individuals (3 and 13–18), ranging in age at imaging from 4 weeks to 17 years of life, were made available and secondarily analyzed by an independent pediatric neuroradiologist (Fig. 3). Analysis included biometry of the corpus callosum for all individuals except for patient 13 (as a digital ruler was not provided with the images) and comparison with age-matched reference values7 (Table S5). Though a spectrum of findings was noted, overall there appeared to be a generalized paucity of white matter with or without white matter T2 hyperintensities. Of note was marked symmetry to the hemispheres of all the children examined with a shallow pattern to the gyri. In addition, the splenium and body of the corpus callosum were generally thin, with varying degrees of dysmorphism.

Brain abnormalities encountered in patients with pathogenic variants in USP7. Age at imaging ranged from full-term corrected to 17 years of life. Though a spectrum of findings was found, the splenium appeared smaller in all children with varying degrees of thinning and dysmorphism to the body of the corpus callosum. Overall, there appeared to be a generalized paucity of white matter with or without white matter T2 hyperintensities. Of note was marked symmetry to the hemispheres of all the children examined with a shallow pattern to the gyri. (a) Normal magnetic resonance image (MRI) for comparison. (b) Patient 3 (at 8 years). (c) Patient 13 (at 31 months). (d) Patient 14 (at 17 months). (e) Patient 15 (at 5 years). (f) Patient 16 (at 27 months). (g) Patient 17 (at 34 weeks’ gestation + 4 weeks of life). (h) Patient 18 (at 17 years).
Variation in head size and shape
No two patients presented with identical MRI findings. Of the seven patients with available scans, four had dysmorphic head shape. Patient 16 (27 months at time of scan; Fig. 3f) showed mild brachycephaly, patient 14 (17 months at time of scan; Fig. 3d) showed microcephaly, and patient 13 (31 months at time of scan; Fig. 3c) showed signs of plagiocephaly. Conversely, patient 17 (Fig. 3g) showed minor macrocephaly (34 weeks’ gestation + 4 weeks of life at time of scan).
Shallow gyri patterns
Gyral patterns were notably simplified in patient 14 and patient 17. Of note was a lack of gyral folding in the gyri of patient 17. Patient 3 (8 years of age at time of scan; Fig. 3b) also had slightly reduced sulci and gyri, but not to the degree of patients 14 and 17.
Symmetry of brain hemispheres
All seven patients scanned had notable symmetry between left and right hemispheres. Symmetry is more evident in some patients than in others.
Dysmorphic ventricular system
In addition to external brain structures, structural abnormalities were identified internally as well. In patients 3, 13, and 16, subtle abnormalities in the shape of the ventricular systems were found. Patient 3 showed slightly enlarged lateral ventricles generally, patient 13 showed an enlarged third ventricle, and patient 16 had slightly dysmorphic frontal horns.
General paucity of white matter, impacting corpus callosum
White matter paucity is the most common MRI finding in patients with USP7 pathogenic variants. Corpus callosum size is a reliable way to judge general myelination and presence of white matter throughout the brain.8 Corpus callosum fronto-occipital diameter (FOD), anteroposterior diameter (APD), genu thickness (GT), isthmus thickness (IT), splenium thickness (ST), body thickness (BT), and APD/FOD ratio were measured following procedures described elsewhere and compared with age-matched reverence values.7 Note that data for patient 18 (17 years of age at time of scan) are compared with reference values for 15-year-old-children because reference values for older children are not available. APD was significantly smaller in this cohort, with a mean Z-score ± standard deviation of −1.16 ± 1.17 (p = 0.0144). Likewise, ST and BT were significantly decreased, showing Z-score means of −0.98 ± 0.82 (p = 0.0034) and −1.18 ± 0.63 (p < 0.0001), respectively. FOD also trended small but comparison with reference values did not reach statistical significance.7 Of note, Z-scores fell more than two standard deviations below the mean for FOD, APD, and BT measures in patient 16, and for APD and IT measures in patient 18 (17 years of age at time of scan; Fig. 3h), indicating severe corpus callosum dysmorphism. The FOD measurement for patient 14 was also more than two standard deviations below the mean.7
Additionally, patient 3, patient 13, and patient 15 (5 years of age at time of scan; Fig. 3e) all had larger total white matter tracts in the corpus callosum. However, unlike normal corpus callosum anatomy (Fig. 3a), the splenium was smaller than, or equal to, the size of the body and genu of the corpus callosum.
White matter hyperintensities, including patterns resembling leukodystrophy
Three of seven patients showed significant white matter hyperintensities. Patient 14 showed multifocal white matter hyperintensities in both frontal and occipital/parietal lobes. Patients 17 and 18 both had frank signal abnormalities that appeared to look much like leukodystrophy, though patient 18 has an additional variant of TMEM106B, pathogenic variants in which are known to cause hypomyelinating leukodystrophy.9 These hyperintensities were the most widespread internal structural abnormalities noted in the scans.
DISCUSSION
Here, we expand and delineate the physical, cognitive, and behavioral phenotypic spectrum of 16 new and 7 previously reported individuals with heterozygous de novo pathogenic variants of the gene USP7. The consistency of clinical features among all individuals, regardless of variant type, supports the pathogenicity of these variants and haploinsufficiency as a pathomechanism for USP7-related disorders, and suggests that reverse phenotyping is possible. Compared with our previously reported findings, the present study confirms the high prevalence (>50%) of DD/ID, ASD, and hypotonia, but shows that seizures and hypogonadism are relatively less common (<50%). Here we also clarify the characteristic behavioral profile and expand the phenotype to encompass speech delays including a nonverbal phenotype, feeding difficulties, GERD, and eye anomalies (such as myopia, estropia, strabismus, and nystagmus). We also broaden the phenotype to include white matter changes on brain MRI, including paucity of white matter, thinning of the corpus callosum, and occasional T2 hyperintensities.
USP7 has been shown to regulate the ubiquitination of a variety of proteins with varying functional diversity. Indeed, USP7 has been implicated in transcription, DNA repair, immune response, and cancer, including the MDM2-p53 mediated pathway.2,3 Interestingly, conditional knockout of Usp7 in the mouse brain resulted in neonatal lethality.10 Moreover, this lethality was not rescuable by the knockout of p53, suggesting a p53-independent neural function. To date, no individuals homozygous null for USP7 have been identified, further supporting a putative critical neuronal function of USP7.
Previously, we reported USP7 to incorporate into the MAGEL2-TRIM27 ubiquitin ligase complex. This complex allows for USP7 to maintain optimal activity of the WASH complex, endosomal F-actin levels, and subsequent protein recycling.1 Importantly, the modification of any of these complexed molecular interactors leads to alterations in proper function of endosomal protein recycling. Indeed, TRIM27 was previously implicated in ASD, while truncating variants in the paternal copy of the maternally imprinted gene MAGEL2 lead to Schaaf–Yang syndrome (SHFYNG), a novel neurodevelopmental disorder.11,12 Moreover, absence of a larger paternally expressed region that contains MAGEL2, 15q11-q13, causes another neurodevelopmental disorder, Prader–Willi syndrome (PWS).13 The clinical phenotypes associated with USP7 variants show marked overlap with those of both SHFYNG and PWS; all show a high prevalence of DD/ID, hypotonia, and feeding difficulties, as well as dysmorphic facial features, eye anomalies (including estropia and myopia), and small hands and feet.12,13,14,15 Though the major diagnostic criteria for PWS delineate the characteristic facial features as including almond-shaped eyes, a small-appearing mouth with downturned corners and a thin upper lip, and dolichocephaly in infancy,15 dysmorphic facial features in SHFYNG and USP7-related disorders are variable and inconsistent.
Despite the overlapping characteristics, each disorder remains distinct. Common features specific to individuals with USP7 pathogenic variants include a characteristic behavioral phenotype and brain MRI anomalies including a paucity of white matter. Furthermore, these individuals do not display excessive weight gain at a prevalence as high as in PWS (16% vs. 66.7%) and do not seem to typically exhibit the hyperphagia present in 84.4% of individuals with PWS, nor do they manifest ASD at a prevalence as high as in SHFYNG (53% vs. 77%); likewise, these individuals also display a lower prevalence of hypogonadism compared with both PWS and SHFYNG (44% vs. 51% vs. 73%) (refs. 12,15). Moreover, of particular note, while the prevalence of contractures in individuals with SHFYNG exceeds 80% (ref. 12), only 25% of individuals with USP7 variants (4/16, two with a deletion and two with a nonsense variant) manifested contractures. Furthermore, though all three disorders are associated with speech delays,16,17,18 the prevalence of a nonverbal phenotype appears greater in SHFYNG (43%) and in USP7-related disorders (18%) than in PWS.
As noted, 11 of the 15 individuals harboring a pathogenic variant in USP7 who had an MRI performed manifested abnormal brain morphology consisting of a paucity of white matter and alterations to gross brain structure. Globally, brain morphology is affected in the forms of micro- and brachycephaly, simplified gyral patterns, and brain hemisphere symmetry. Specific findings included a paucity of white matter in deep and subcortical regions, thinning of the corpus callosum, and abnormally shaped ventricular systems of the central nervous system.
As with USP7 pathogenic variants, PWS is likewise associated with ventriculomegaly19 and white matter changes,20,21,22 including white matter lesions.23 Biometry measurements are not available for comparison, though interestingly, altered white matter microstructure (measured by fractional anisotropy) has been noted in PWS areas including the corpus callosum21 and specifically in the splenium.22 However, rather than simplified or absent gyral folding, polymicrogyria has been found in individuals with PWS.19 In addition, though corpus callosum thinning is characteristically caused by genetic and metabolic disorders as well as disruptions in neurodevelopment,8,24 agenesis/hypoplasia of the corpus callosum is a relatively uncommon (<3%) finding in PWS.25 Thus, despite some overlap, the constellation of MRI findings associated with USP7 pathogenic variants differs from that of PWS. Though formal evaluation of brain morphology in SHFYNG has not yet been conducted, preliminary assessment of patients with SHFYNG show that white matter abnormalities are not evident to the degree seen in patients with USP7 variants. It appears that abnormalities in white matter help further distinguish the phenotype of USP7 variants from that of truncating variants in MAGEL2 causal to SHFYNG.
While pathogenic variants of USP7 are consistently correlated with white matter and corpus callosum anomalies identified via MRI, along with the physical and behavioral characteristics described above, phenotype-first diagnosis remains unlikely. Ascertainment bias, unstandardized phenotyping, a small sample size, and varying patient ages are limitations for the findings presented here. Furthermore, an individual’s age and IQ can affect whether certain potential diagnoses and features are relevant, such as ASD, characteristic behaviors, and hypogonadism. Additionally, while it is plausible that patient 18’s USP7 variant led to his white matter and corpus callosum anomalies, the possibility of his TMEM106B variant contributing to these MRI findings should also be considered, given this gene’s association with hypomyelinating leukodystrophy.9 Whether the variant in TMEM106B acts in conjunction with the loss of USP7 function has yet to be explored. Furthermore, patient 20 harbors an additional de novo pathogenic variant in SLC2A1, associated with several overlapping features including DD/ID, absent speech, seizures, and aggressive behavior,26 likewise raising the possibility of a dual diagnosis or multiple-variant compound effects.
Proper assessment of patients with complicated neurologic and physiological abnormalities including hypotonia, DD/ID, ASD, hypogonadism, and speech delay can be difficult, considering the phenotypic overlap with other known neurodevelopmental disorders like PWS and SHFYNG. USP7-associated neurodevelopmental disorder represents a novel genetic syndrome with features similar to both SHFYNG and PWS. Defining features of individuals with pathogenic variants/deletions of USP7 include severe speech delay, unique MRI anomalies, seizures in 48% of individuals, and a lack of high prevalence of the contractures typically observed in SHFYNG. Characteristic MRI findings (e.g., white matter paucity, corpus callosum thinning, etc.) may provide a key piece of evidence to raise clinical suspicion of USP7-associated neurodevelopmental disorder.
References
Hao Y-H, Fountain MD, Fon Tacer K, et al. USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol Cell. 2015;59:956–969.
Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653.
Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60:61–68.
Schaefer JB, Morgan DO. Protein-linked ubiquitin chain structure restricts activity of deubiquitinating enzymes. J Biol Chem. 2011;286:45186–45196.
Hao Y-H, Doyle JM, Ramanathan S, et al. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell. 2013;152:1051–1064.
Gomez TS, Billadeau DD. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. 2009;17:699–711.
Garel C, Cont I, Alberti C, Josserand E, Moutard ML, Ducou le Pointe H. Biometry of the corpus callosum in children: MR imaging reference data. AJNR Am J Neuroradiol. 2011;32:1436–1443.
Walterfang M, Fahey M, Abel L, et al. Size and shape of the corpus callosum in adult Niemann-Pick type C reflects state and trait illness variables. AJNR Am J Neuroradiol. 2011;32:1340–1346.
Simons C, Dyment D, Bent SJ, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 2017;140:3105–3111.
Kon N, Zhong J, Kobayashi Y, et al. Roles of HAUSP-mediated p53 regulation in central nervous system development. Cell Death Differ. 2011;18:1366–1375.
St Pourcain B, Whitehouse AJO, Ang WQ, et al. Common variation contributes to the genetic architecture of social communication traits. Mol Autism. 2013;4:34
Fountain MD, Aten E, Cho MT, et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet Med. 2017;19:45–52.
Schaaf CP, Gonzalez-Garay ML, Xia F, et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat Genet. 2013;45:1405–1408.
Butler MG, Meaney FJ. An anthropometric study of 38 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. 1987;26:445–455.
Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108:E92.
McCarthy J, Lupo PJ, Kovar E. et al. Schaaf-Yang syndrome overview: report of 78 individuals. Am J Med Genet A. 2018;176:2564–2574.
Defloor T, Van Borsel J, Curfs L. Articulation in Prader–Willi syndrome. J Commun Disord. 2002;35:261–282.
Lewis BA. Speech and Language Disorders Associated with Prader-Willi Syndrome. In: Butler MG, Lee PDK, Whitman BY, eds. Management of Prader-Willi Syndrome. New York: Springer. 2006; 272–283.
Miller JL, Couch JA, Schmalfuss I, He G, Liu Y, Driscoll DJ. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in Prader–Willi syndrome. Am J Med Genet A. 2007;143A:476–483.
Xu M, Zhang Y, von Deneen KM, Zhu H, Gao J-H. Brain structural alterations in obese children with and without Prader-Willi syndrome. Hum Brain Mapp. 2017;38:4228–4238.
Lukoshe A, van den Bosch GE, van der Lugt A, Kushner SA, Hokken-Koelega AC, White T. Aberrant white matter microstructure in children and adolescents with the subtype of Prader-Willi syndrome at high risk for psychosis. Schizophr Bull. 2017;43:1090–1099.
Yamada K, Matsuzawa H, Uchiyama M, et al. Brain developmental abnormalities in Prader-Willi syndrome detected by diffusion tensor imaging. Pediatrics. 2006;118:e442–e448.
Miller J, Kranzler J, Liu Y, et al. Neurocognitive findings in Prader-Willi syndrome and early-onset morbid obesity. J Pediatr. 2006;149:192–e3.
Putoux A, Nampoothiri S, Laurent N, et al. Novel KIF7 mutations extend the phenotypic spectrum of acrocallosal syndrome. J Med Genet. 2012;49:713–720.
Torrado M, Foncuberta ME, Perez MF, et al. Change in prevalence of congenital defects in children with Prader-Willi syndrome. Pediatrics. 2013;131:e544–e549.
Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. 2013;13:342.
Acknowledgements
This study was partially supported by Intellectual and Developmental Disabilities Research Center (IDDRC) grant 1U54HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, and by a grant to J.W. from the National Science Centre of Poland, project UMO-2014/15/B/NZ5/03503. Funding was also provided by the German Federal Ministry of Education and Research (BMBF) (CHROMATIN-Net 01GM1520C to F.J.K.), and the Medical Faculty of the University of Lübeck (J09-2017 to I.P.). We acknowledge HUGODIMS consortium, supported by a grant from the French Ministry of Health and from the Health Regional Agency from Poitou-Charentes (HUGODIMS, 2013, RC14_0107); we are grateful for the support of Frédérique Allaire from the Health Regional Agency of Poitou-Charentes. We appreciate the support of Kym Boycott, from the Children’s Hospital of Eastern Ontario and the Children’s Hospital of Eastern Ontario Research Institute, and Taila Hartley, from the Children's Hospital of Eastern Ontario Research Institute. We are grateful to Charles Minard from Baylor College of Medicine for his discussion and input. The National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under award numbers U01HG007672 (Duke University) and U01HG007942 (Baylor College of Medicine-Sequencing) also supported this research. The content is solely the responsibility of the authors and does not necessarily represent the NIH’s official views. We thank the Undiagnosed Disease Network (UDN) and the iHope Program, supported by Illumina, for each diagnosing a patient reported. UDN members are listed in supplementary materials. Finally, the authors are indebted to the patients and their families for their participation in this study. A Facebook group for affected families can be found at https://www.facebook.com/groups/969399453182041/. Web pages for the Foundation for USP7-Related Diseases (USA) and the Association Manger la Vie (France) can be found at www.usp7.org and http://mangerlavie.wixsite.com/association, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
E.T., J.J., F.M., I.M.W., and R.E.P. are employees of GeneDx, Inc., a wholly owned subsidiary of OPKO Health, Inc. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories, including chromosomal microarray and exome sequencing. The other authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Fountain, M.D., Oleson, D.S., Rech, M.E. et al. Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies. Genet Med 21, 1797–1807 (2019). https://doi.org/10.1038/s41436-019-0433-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0433-1
