
Abstract
Purpose
To enhance classification of variants of uncertain significance (VUS) in the DNA mismatch repair (MMR) genes in the cancer predisposition Lynch syndrome, we developed the cell-free in vitro MMR activity (CIMRA) assay. Here, we calibrate and validate the assay, enabling its integration with in silico and clinical data.
Methods
Two sets of previously classified MLH1 and MSH2 variants were selected from a curated MMR gene database, and their biochemical activity determined by the CIMRA assay. The assay was calibrated by regression analysis followed by symmetric cross-validation and Bayesian integration with in silico predictions of pathogenicity. CIMRA assay reproducibility was assessed in four laboratories.
Results
Concordance between the training runs met our prespecified validation criterion. The CIMRA assay alone correctly classified 65% of variants, with only 3% discordant classification. Bayesian integration with in silico predictions of pathogenicity increased the proportion of correctly classified variants to 87%, without changing the discordance rate. Interlaboratory results were highly reproducible.
Conclusion
The CIMRA assay accurately predicts pathogenic and benign MMR gene variants. Quantitative combination of assay results with in silico analysis correctly classified the majority of variants. Using this calibration, CIMRA assay results can be integrated into the diagnostic algorithm for MMR gene variants.
Similar content being viewed by others

INTRODUCTION
Lynch syndrome (OMIM 120435), a common hereditary predisposition to colorectal and other cancers, is caused by a dominantly inherited defect in one of four genes involved in postreplicative DNA Mismatch Repair (MMR): MLH1 (OMIM 120436), MSH2 (OMIM 609309), MSH6 (OMIM 600678), and PMS2 (OMIM 600259). Somatic loss of the wild-type (WT) allele results in cellular MMR deficiency. The resulting inability to correct errors by the replicative DNA polymerases is considered the critical mechanism leading to Lynch-associated cancers, by causing a spontaneous “mutator phenotype” in affected cells.1
Determining pathogenicity of the increasingly prevalent variants of uncertain significance (VUS) in cancer-predisposing genes provides a major challenge to clinical geneticists.2,3,4,5,6 Currently, the large majority of missense variants identified in MMR genes cannot be classified.7,8,9,10,11,12,13 As of March 2018, ~94% of the MMR gene missense variants listed in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) lack clinically useful classifications, emphasizing the need for improved classification methods.12
Commonly used methods to analyze missense variants in MMR and other cancer predisposition–associated genes include sequence analysis-based in silico prediction, segregation in families, population allele frequencies, and tumor pathology.7,9,10,11,12 The International Society for Gastrointestinal Hereditary Tumors (InSiGHT) Variant Interpretation Committee (VIC) has evaluated qualitative or quantitative integration of evidence to classify variants (https://www.insight-group.org/)12 using standards set by the International Agency for Research on Cancer (IARC).8 The VIC has reclassified a limited number of MMR gene VUS as clinically pathogenic (IARC class 5, probability of pathogenicity >0.99; plus IARC class 4, probability of pathogenicity >0.95) or as clinically benign (IARC class 1, probability of pathogenicity <0.001); or IARC class 2 (probability of pathogenicity <0.05) (ref. 12) with associated clinical recommendations.8,13 Despite these efforts, current strategies to assess and combine different types of evidence are inadequate, leaving most VUS as IARC class 3, Uncertain,8,12 often due to insufficient clinical data.
It is felt that biochemical assays of MMR function could strongly contribute toward classifying MMR gene VUS.13,14,15 A central challenge in adding a new data type, such as a biochemical assay, to a variant classification system, is calibration of the new assay; that is, conversion from the natural output of the assay to the weighting units used by the classification system. This is true whether systems are fundamentally qualitative, with weightings set by expert opinion,12,13 or largely quantitative with various evidence types fitted to a common numeric scale.7,10,16,17,18 Quantitative evaluation of BRCA and MMR gene variants has used naive Bayesian classifiers, where the unit systems are probabilities in favor of pathogenicity, usually expressed as a prior probability (Prior-P) and either odds in favor of pathogenicity (OddsPath) or likelihood ratios in favor of pathogenicity.7,10,19 To be confident in this method, a validation step should demonstrate that the calibration is accurate and reproducible.
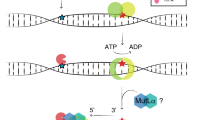
We developed the cell-free in vitro MMR activity (CIMRA) assay, a biochemical test that only requires variant sequence data to assess missense and small indel variants and can be performed in a few days using common laboratory equipment (Fig. 1a, Figure S1) (refs 20,21,22). Here, we calibrate the assay by converting output into OddsPath and probabilities, and validate it using a preplanned symmetric training and cross-validation strategy assessing reproducibility of point estimates and confidence intervals (CIs) obtained from two independent training runs (Fig. 1b).23

Outline of the cell-free in vitro MMR activity (CIMRA) assay and of this study. (a) Outline of the CIMRA assay. The figure describes testing of an MSH2 variant but testing MLH1 variants is technically similar. (b) Flow diagram for major steps of the study design. The variants in the "CIMRA Assays set 1 and 2" boxes are coloured according to their InSiGHT classification, see also Fig. 2. PCR polymerase chain reaction, ROC receiver operating characteristic, WT wild type.
MATERIALS AND METHODS
Selection of classified missense substitutions for the calibration study
In January 2014, we reviewed the InSiGHT variant database (http://insight-group.org/variants/database/) for all MLH1 and MSH2 missense substitutions that met InSiGHT criteria as pathogenic (IARC classes 5 or 4) or benign (IARC classes 1 or 2) (refs. 8,12). Among the classified missense substitutions, 86 had been assigned to one of these classes without need for functional assay results (62 were class 4/5, and 24 were class 1/2). To pick substitutions for the first training set, we made a random draw of 35 substitutions from the class 4/5 missense substitutions and, separately, 15 from the class 1/2 missense substitutions.
In December 2014, we re-reviewed the MLH1 and MSH2 missense substitution classifications in the InSiGHT database. Due to changes in classification during 2014, there were 58 missense substitutions in this list, not in the first training set of 50, that were classified without need for functional assay results. To pick substitutions for the second round of CIMRA assays, we used a random draw to drop 8 from this group of 58, leaving n = 50 (n = 37 class 4/5 and n = 13 class 1/2) substitutions.
Before completing analysis of the CIMRA assay data, we recognized that 30 of the 100 substitutions had previously been used to calibrate sequence analysis–based computational algorithms, and functional assay results may have been required for selection of some of those variants for that study.11 To prevent potential bias in CIMRA assay calibration, and to improve independence from previous calibration of key computational algorithms, these 30 variants were excluded from the CIMRA assay training. This left 35 substitutions (25 class 4/5 and 10 class 1/2) in training set 1, and 35 substitutions (again, 25 class 4/5 and 10 class 1/2) in training set 2.
In vitro MMR activity (CIMRA) assays
Complete MMR gene complementary DNAs (cDNAs) were cloned in the pCITE4a plasmid and used as a template for polymerase chain reaction (PCR) to recreate variant alleles including the vector-derived T7 promoter and CITE sequences that are required for efficient transcription/translation in vitro, as described.20,21,22 Mutagenic oligonucleotides (sequences available upon request) were ordered from Integrated DNA Technologies. PCR reactions were performed with Pfx Platinum Polymerase (Invitrogen). Proper introduction of the variant codon was confirmed by direct sequencing. Control (wild-type) MMR genes were obtained from parallel PCR reactions using primers containing silent mutations. PCR products were purified using the PCR Purification Kit (Qiagen) and used for in vitro expression of variant proteins in the TNT Quick Coupled Transcription/Translation System (Promega), in the presence of PCR Enhancer and Fluorotect, with minor modifications to the manufacturer’s protocol. Protein expression levels were verified by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and fluorescent imaging (Typhoon 9410, GE Healthcare). Wild-type heterodimeric partners of the variant proteins were produced by large-scale in vitro expression from the respective MMR genes cloned in pCITE4a. Heterodimeric variant MSH2/MSH6 or MLH1/PMS2 proteins were generated by allowing the partners (1:1 v/v) to dimerize with their variant for 15 minutes at room temperature.
CIMRA assays were performed essentially as described.20,21,22 Nuclear extracts were prepared as described,24 either from HCT116 cells lacking PMS2/MLH1 or from LoVo cells lacking MSH2/MSH6. Extracts were complemented with TNT expression mix containing variant or wild-type PMS2/MLH1 or MSH2/MSH6, respectively, and fluorescent G:T-mismatched substrate pJHGT3’lnFAM (prepared as described).20,21,22 After incubating 40 minutes at 37 °C, substrate DNA was extracted with phenol/chloroform and ethanol-precipitated. The substrate was digested with HinDIII, which uniquely cleaves repaired substrate, and with BsrBI (Fermentas) to cleave at both sides of the (former) mismatch. Digested substrate was mixed with Hi-Di Formamide and GeneScan-500 ROX size standard (Applied Biosystems). Fluorescent fragment analysis was performed under standard conditions using an Applied Biosystems 3100 Genetic Analyzer. Signals were quantified using GeneMarker software (Softgenetics). Repair levels were calculated as the ratio between the height of the repair-specific peak and the total fluorescent signal, normalizing to the activity of the respective wild type. All variants were tested at least three times, using independently generated and expressed PCR products. Each included established MMR-deficient controls: p.G67R for MLH1 (refs 20,25) and p.A636P for MSH2 (refs 21,26).
Regression for CIMRA assay calibration
For CIMRA assay calibration regressions, InSiGHT observational data in favor of pathogenicity in the form of Log10(OddsPath) was treated as the dependent variable. The normalized CIMRA assay value for the substitutions was treated as an independent variable, and gene symbol was coded as an indicator variable. We performed multivariate linear regressions on Log10(OddsPath) versus CIMRA assay value, enabling estimation of CIMRA OddsPath from assay values. We calculated Working–Hotelling 80% and 95% confidence band limits to some of the key regressions.27
For receiver operating characteristic (ROC) area under the curve (AUC) analyses, we simplified data into two classifications, collapsing the InSiGHT qualitative classes 5/4 into “pathogenic” and classes 1/2 into “benign.” This binary classification was used as the reference variable and CIMRA assay values were used as the classification variable. Assay calibration regressions and ROC AUC analyses were performed in Stata/SE 15.0 (StataCorp).
In silico analyses and Bayesian integration
In silico analyses predicting the probability of pathogenicity for each variant were performed using the programs MAPP and PolyPhen2 as previously reported.11 We used these values as the computational Prior-P for further analyses. We set upper and lower limits for in silico prior probability values at 0.10 and 0.90 (ref. 11). Results could then be integrated, using Bayes’ rule, with other data for which OddsPath can be determined (e.g., segregation, tumor pathology), to obtain posterior probabilities of pathogenicity (Post-P),10 hereafter termed “two-component” analysis or “two-component Post-P.” For ROC AUC analyses, the binary InSiGHT classification defined above was used as the reference variable and two-component Post-P as the classification variable.
We estimated a global Prior-P for MLH1 and MSH2 missense variants by scoring all missense substitutions in these genes recorded in either the InSiGHT database (N = 773) or gnomAD (N = 1116) as of 11 April 2018, using the programs MAPP and PolyPhen2 as previously reported.11 For the subset that were either unclassified or explicitly class 3 (N = 1299), the average Prior-P was 0.39 (Table S1).
Sensitivity and specificity at the classification thresholds
For estimations of sensitivity and specificity of the CIMRA assay alone, CIMRA OddsPath were converted to probabilities of pathogenicity using Bayes’ rule, at a Prior-P of 0.39. Missense substitutions with CIMRA probability of pathogenicity >0.95 were considered “predicted class 4/5,” and those with CIMRA probability of pathogenicity <0.05 were considered “predicted class 1/2.”
For sensitivity and specificity of “CIMRA predicted class 4/5,” true positives were InSiGHT class 4/5 variants with CIMRA probability of pathogenicity >0.95, and true negatives were InSiGHT class 1/2 variants with CIMRA probability of pathogenicity ≤0.05. Sensitivity was estimated as (# true positives)/(# InSiGHT class 4/5 variants). Specificity was estimated as (# true negatives)/(# InSiGHT class 1/2 variants).
For sensitivity and specificity of “CIMRA predicted class 1/2,” true positives were InSiGHT class 1/2 variants with CIMRA probability of pathogenicity <0.05, and true negatives were InSiGHT class 4/5 variants with CIMRA probability of pathogenicity ≥0.05. Sensitivity was estimated as (# true positives)/(# InSiGHT class 1/2 variants). Specificity was estimated as (# true negatives)/(# InSiGHT class 4/5 variants).
After Bayesian integration of the computational Prior-P with CIMRA OddsPath, substitutions with a two-component Post-P >0.95 were considered “two-component class 4/5,” and those with a two-component Post-P <0.05 were considered “two-component class 1/2.” Sensitivity and specificity of two-component class 4/5 and class 1/2 were then re-estimated as above.
Cell-based assays
The introduction of variants in MLH1 (vector pCite-MLH1) and MSH2 (vector pAL112) (primer sequences are available upon request) was performed according to the manufacturer’s protocol (QuikChange II XL, Agilent Technologies). Substitutions were verified by sequencing.
Plasmid pCite-MLH1 (wild type or mutant) was digested with NdeI and EcoRI and inserted into yeast two-hybrid (Y2H) vector pGBKT7 (bait, Clontech). MSH2 variants were inserted into the NcoI site in pGK240. The coding sequences were subcloned into two-hybrid vector pGBKT7. PMS2 was inserted in NdeI and SmaI sites of vector pGK239 (pGADT7 backbone, prey). MSH6 was subcloned from pGEX-MSH6 into pGADT7.
Y2H analysis was set up according to the manufacturer’s protocol (Clontech). The Y2HGold yeast strain was cotransformed with PMS2/MLH1- or MSH2/MSH6-expressing plasmids. Plates were grown for 3–5 days. The PMS2 wild type, MLH1 wild type, and MLH1 mutants were plated on plates lacking Trp/Leu and containing X-α-gal, and plates lacking Trp/Leu/His/Ada and containing X-α-gal and Aureobasidin A. The MSH6 wild type, MSH2 wild type, and MSH2 mutants were plated on plates lacking Trp/Leu and containing X-α-gal and on plates lacking Trp/Leu/His and containing X-α-gal and Aureobasidin A. Colonies were counted and scored for growth and for blue (interaction)/white (no interaction).
Subcellular localization
Subcellular localization was assayed as described with few modifications.28 In brief, wild-type MLH1 and MSH2 were subcloned into pEYFP (Contech). Wild-type and variant-bearing MLH1 and MSH2 constructs were subcloned into pECFP1 (Clontech). Murine NIH3T3 fibroblasts were maintained in Dulbecco's Modified Eagle's medium (DMEM) glutamax with 1% penicillin–streptomycin and 10% fetal bovine serum (FBS). Cells were seeded onto glass coverslips in 6-well glass bottom dishes one day prior to transfection and transiently cotransfected using PolyJet reagent (SignaGen) with: For MSH2: pECFP-MSH2 VUS or pECFP-MSH2 WT together with pEYFP-MSH2 WT. For MLH1: pECFP-MLH1 VUS or pECFP-MLH1 WT together with pEYFP-MLH1 WT. Cells were incubated overnight at 37 °C, humidified, with 5% CO2. Twelve to eighteen hours after transfection, cells were formaldehyde-fixed and mounted onto a glass slide with VectaShield mounting medium (H-1000). Subcellular localization of fusion proteins was analyzed using Nikon Eclipse 80i equipped with NIS-Elements AR software, with pictures processed using ImageJ.
Multilaboratory assessment of CIMRA assay reproducibility
Variants for multilaboratory assessment of CIMRA assay reproducibility were selected manually, based on differential repair activities and differential positions in the MSH2 and MLH1 proteins.
CIMRA assays were performed as described above, with minor modifications. Substrate pJHGT3’lnFAM was modified to substrate pYT3c in which the G·T mismatch is embedded in a Hin1II site rather than a HinDIII site. Because two additional Hin1II sites exist in the vector backbone, repair activity can be quantified using one restriction enzyme rather than two. Also, cell extracts were prepared from HeLa cells that were made either MSH2/MSH6 deficient (for MSH2 variants) or MLH1 deficient (for MLH1 variants) using CRISPR/Cas9. Verification of in vitro protein expression was omitted.
Reagents (e.g., buffer and cell extract–containing CIMRA mix, substrate plasmid, templates for MSH2 and MLH1 PCRs, MSH6 and PMS2 expression plasmids) were prepared in large quantities at Leiden University Medical Center (LUMC) and were distributed from single batches to participating labs by mail (Figure S2). Commercially available components (e.g., TNT Quick Coupled Translation Kit, restriction enzymes, PCR polymerase, primers, etc.) were ordered by participating labs, which received a detailed protocol and troubleshooting support by email. Results were not shared among laboratories until all experiments were completed.
Leave-one-out cross-validation was used to assess reproducibility of each lab separately. As above, InSiGHT Log10(OddsPath) was treated as dependent variable and CIMRA assay results from each laboratory as an independent variable in a linear regression. The predicted Log10(OddsPath) was used to categorize the single holdout variant from each regression as likely pathogenic, unclassified, or likely neutral, with category boundaries as described above. The predicted class was cross-tabulated against InSiGHT class. The cvTools package in R version 3.2.1 (ref. 29) was used for cross-validation.
RESULTS
CIMRA assay training, cross-validation, and calibration
To integrate CIMRA assay results with other quantitative data,7,12 we applied a symmetric training and cross-validation strategy (Fig. 1b). In the first training cycle, the CIMRA assay was used to determine MMR activity of 35 variants previously classified by InSiGHT without requiring functional assay data (Fig. 2a). Variants previously used to calibrate the computational Prior-P were also excluded from training because their selection for that study often depended on functional assays.11 This would introduce a circularity by evaluating the CIMRA assay using variants whose function in a prior assay was necessarily concordant with their classification.11 CIMRA assay results from the first training set had a ROC AUC of 0.94, concordant with their previously assigned class for 33/35 variants (Fig. 2b). Regression of these training data against the OddsPath from InSiGHT patient observational data resulted in an initial calibration equation, graphed in Fig. 2c and detailed in Table S3.

Calibration and validation of the CIMRA assay. (a) Relative repair efficiencies for MLH1 and MSH2 variants from the first CIMRA training set. Variants are colored according to their InSiGHT classification (see legend in figure). The MSH2 p.A636P variant and the MLH1 p.G67R variant are included in every experiment as repair-deficient (pathogenic) controls. Bars represent mean ± SEM of 3–5 experiments. Asterisks indicate variants whose MMR activity appears discordant with their original InSiGHT classification. (b) Receiver operator characteristic (ROC) curve for the first CIMRA assay training. (c) Regressions of first (blue) and second (red) CIMRA assay training values against odds in favor of pathogenicity. Both curves are embedded in their 80% confidence envelopes. Note that the y-axes in (c) and (f) display probability of pathogenicity rather than Log(odds in favor of pathogenicity), to emphasize sigmoid calibration bounded at probabilities of 1.00 and 0.00. (d) As in (a), but for second CIMRA assay training. (e) ROC curve for the second CIMRA assay training. (f) As in (c), but shown here is the final calibration curve combining first CIMRA assay training (n = 35) and second CIMRA assay training (n = 35) substitutions; the regression curve is embedded in both 80% and 95% confidence envelopes. IARC International Agency for Research on Cancer.
CIMRA assay training was repeated using a second, independent, set of 35 variants (Fig. 2d). The CIMRA assay results were perfectly concordant with their InSiGHT classifications (ROC AUC = 1.00, Fig. 2e). Defining “true positive” as class 4/5, in cross-validation between the two CIMRA training runs, the specificity was 1.00 with a sensitivity of 0.65. Redefining “true positive” as prediction of a neutral variant as neutral, the specificity of class 1/2 was 0.96 with a sensitivity of 0.65. Importantly, the regression equations resulting from the first and second CIMRA training sets were concordant with each other; i.e., the slope and intercept point estimates from one fell within the corresponding 80% confidence intervals of the other, and vice versa, meeting our predefined assay cross-validation criterion (Fig. 2c, Table S3, Figure S3 [ref. 23]).
Concordance between the two training regressions justified combining the two data sets for a final calibration equation (1):
(Fig. 2f, Table S3). This equation converts CIMRA assay activity into CIMRA OddsPath, the variable required for Bayesian integration with other data to generate a posterior probability of pathogenicity that can classify variants for clinical recommendations.7,8,10,11,12,18
After our experiments were completed, InSiGHT reclassified two variants with intermediate CIMRA assay activity from class 4 to class 3 (MSH2 p.R524L and MLH1 p.N64S); these were omitted from analyses beyond this point. Using results from the calibrated CIMRA assay alone, ROC AUC was 0.980 (95% CI = 0.95–1.0; Fig. 3a). The specificity of class 4/5 was again 1.00 with a sensitivity of 0.60. The specificity of class 1/2 was 0.96 with a sensitivity of 0.75. The calibrated CIMRA assay was able to correctly classify 65% (n = 44/68) of variants, with only two discordant classifications (MLH1 p.A681T and MSH2 p.P652H); 32% (n = 22/68) of variants remained in class 3 (Table S2).

A Bayesian integration of cell-free in vitro MMR activity (CIMRA) assay values and in silico predictions of pathogenicity correctly classifies ~90% of missense variants. (a) Receiver operating characteristic (ROC) curve of a cross-validation between the two CIMRA training runs. (b) ROC curve of a two-component (CIMRA assay + in silico) analysis. (c) Distribution of probabilities of pathogenicity for 68 missense variants as estimated by in silico analysis only (top), CIMRA only (middle), or integrated two-component analysis (CIMRA plus the computational Prior-P; bottom). The gray, dotted, lines highlight probabilities of 0.05 and 0.95. #: Because the probabilities of pathogenicity estimated by the computational Prior-P are capped at 0.1 and 0.9, the bins <0.08 and >0.92 are empty. (d) Subcellular localization of wild-type (WT) and mutant MLH1 protein. NIH3T3 cells were transiently transfected with pYFP-MLH1 WT (shown in red, left panel) together with pCFP-MLH1 (WT or mutant; shown in green, middle panel). The localization of the fusion proteins were analyzed using confocal laser scanning microscopy. The corresponding outline of cells in Nomarski contrast are shown (right panel) where the yellow color indicates overlay of the two proteins. IARC International Agency for Research on Cancer.
Bayesian integration of CIMRA assay results with the computational Prior-P
We used Bayes’ rule to quantitatively integrate the CIMRA assay-based OddsPath with the previously calibrated computational Prior-P.11 Of 48 variants classified by InSiGHT using clinical data as class 4/5, the resulting two-component analysis (CIMRA plus in silico) classified 43 variants as class 4/5, left 4 variants as class 3, and classified 1 as class 1/2 (MLH1 p.A681T; Fig. 3c and Table S2). Of 20 variants previously classified as class 1/2, the two-component analysis corroborated the assessment of 16 as class 1/2, left 3 variants as class 3, and classified 1 as class 4/5 (MLH1 p.K618T; Fig. 3c, Table S2). The specificity of the two-component classification of class 4/5 was 0.95 with a sensitivity of 0.90. The specificity of classification of class 1/2 was 0.98 with a sensitivity of 0.80. The AUC of the ROC curve was 0.977 (0.95–1.00; Fig. 3b). Thus, a two-component analysis correctly assessed 87% (n = 59/68) of a random set of previously classified MSH2 or MLH1 missense substitutions with a discordance rate of 2.9% (n = 2/68; Table S2). Compared with the CIMRA assay alone, integration of CIMRA assay data with the computational Prior-P reduced the proportion of variants left in class 3 from 32% (n = 22/68) to 10% (n = 7/68) (Fig. 3c), without changing the error rate (2/68 = 2.9%).
Accessory assays further facilitate variant classification
The 2.9% discordant classifications between the clinical data–based (InSiGHT) and our integrated (in silico plus CIMRA) analyses might reflect their incorrect classification by either or both approaches. We explored whether we could resolve these rare discordances, the 10% inconclusive classifications from our integrated approach, and variants whose in silico and CIMRA assay results were discordant, by testing in two accessory assays: (1) subcellular localization using fluorescently tagged proteins, and (2) protein heterodimer formation using the Y2H assay (Table 1).
The phenotypes of the two discordantly classified variants in the accessory assays corroborated their CIMRA assay activities (Fig. 3d, Table 1). MLH1 variant p.A681T (InSiGHT class 4/5, CIMRA activity >70%) was normal in accessory assays; p.A681T displays an expression defect in vivo30 that evidently does not result in a defect in the CIMRA assay. MLH1 p.K618T (InSiGHT class 1, but reduced CIMRA activity of 35%) was defective in the Y2H assay.
CIMRA assay activities and the computational Prior-P were mildly discordant for 3 MLH1 variants. Still, our two-component analysis correctly classified these variants as class 1/2 and, in concordance, these variants displayed wild-type dimerization and localization. All five MLH1 and MSH2 variants with low computational Prior-P but slightly reduced CIMRA assay activities were correctly classified as class 1/2 and displayed normal dimerization and localization. This further emphasizes the robustness of our two-component variant classification. Three variants assigned to class 3 by our two-component analysis displayed a defect in the Y2H assay and/or in intracellular localization, supporting pathogenicity (and their InSiGHT classification; Table 1). Based on these results, we anticipate that accessory assays will help classify the minority of the 10% of VUS remaining in class 3 using our two-component approach.
Interlab CIMRA assay comparison
To further validate the CIMRA assay as a tool in the diagnosis of MMR protein variants, MMR activity of ten MSH2 and ten MLH1 variants, plus internal controls, were tested in five independent laboratories worldwide (Figure S2), using an optimized CIMRA assay protocol (Methods). Using leave-one-out cross-validation to assess assay performance at each of the labs, correct classification was obtained in 90 of 100 events. There were eight events where the CIMRA assay alone would have left a variant in class 3, and two events where the pathogenic variant MSH2 p.L93F (which had CIMRA activity of 50.5% in the main assay calibration study) would have been placed in class 2 (Fig. 4 and Table S4). Thus the multilaboratory assessment of the CIMRA assay showed strong qualitative and quantitative interlaboratory reproducibility for both genes.

Multilaboratory assessment of cell-free in vitro MMR activity (CIMRA) assay reproducibility. (a) MSH2 variants and controls were tested for CIMRA assay activity in different centers worldwide (see legends in figure). The MSH2 p.A636P variant is included in every experiment as a repair-deficient control. Bars represent mean ± SEM of 3–4 experiments. Numbers below the diagrams indicate the IARC classification. * Variant discordantly classified, # variant unclassified. (b) As with (a), but for MLH1 variants, where MLH1 p.G67R is the experimental control. IARC International Agency for Research on Cancer, LUMC Leiden University Medical Center, QIMR QIMR Berghofer Medical Research Institute, UMCG University Medical Center Groningen, WT wild type.
DISCUSSION
Current variant classification guidelines from the American College of Genetics and Genomics envision that “well established in vitro…functional studies supportive of a damaging effect” provide strong evidence of pathogenicity, and that similar studies showing “no damaging effect on protein function” provide strong evidence of benign impact.13 Quantitative modeling indicates that “strong” evidence is consistent with OddsPath between 18.7:1 and 350:1 (ref. 18). Additionally, the IARC 5-class system for sequence variant classification8 accommodates 5% error rates in classes 1/2 and 4/5. Toward efficient reclassification of VUS to class 4/5, the error rates of both CIMRA assay alone and of the two-component classification meet these standards; they are sufficiently robust to reclassify variants with weak clinical data to class 4/5. Toward reclassification to class 1/2, the error rates fall slightly short, consistent with the fact that mechanisms other than loss of MMR function in vitro can lead to pathogenicity, such as altered splicing, or reduced protein expression or half-life, as exemplified by MLH1 p.A681T.
Analysis of variants with intermediate repair capacity suggests that the pathogenicity of variants with CIMRA activity between ~35% and ~50% could be influenced by other cellular factors. The single variant that was classified benign by InSIGHT but had low-intermediate CIMRA activity, MLH1 p.K618T, likely falls into this category. By analogy to MLH1 p.K618A31 and BRCA1 p.R1699Q,32 some variants likely confer moderate risk for Lynch syndrome malignancies. Calibrating CIMRA results as a continuous variable addresses this issue by assigning less extreme odds to variants with intermediate activity, providing a natural weighting for classification, and avoiding “edge effects” that would improperly weight results in the activity interval where CIMRA is essentially indeterminate. For these variants, other data must aid classification. Evolving data-sharing efforts such as ClinVar and ClinGen33 will focus attention on VUS requiring additional clinical or epidemiological data. Future studies can assess intermediate CIMRA activity and penetrance.
Ideally, both variant-level and clinical data are included in variant classification. Thus, we suggest that expert panels that integrate functional data into classification schemes consider setting limits for functional data just shy of the thresholds for class 4 or class 2, so that a small amount of concordant clinical evidence is required to reach these clinically relevant classifications. Similar limits were suggested for calibration of in silico algorithms.11 We emphasize that the ROC curve and error rates of our approach compare favorably with other diagnostic tools used in clinical medicine34 and warrant the integration of CIMRA assay data into the MMR gene variant classification algorithm.
In conclusion, we present the CIMRA assay, as well as our two-component analysis, as a thoroughly calibrated and validated analytical tool that is suited for clinical use to assess the pathogenicity of MMR gene missense variants and small indels in the diagnosis of Lynch syndrome. The calibration described takes strength from three specific distinctions: CIMRA is an in vitro biochemical assay that directly tests the key function1 of MMR proteins, the sets of variants used to calibrate the computational Prior-P11 and the CIMRA assay are nonoverlapping, and CIMRA assay calibration met a predefined, peer reviewed,23 validation criterion. Given the hurdles in collecting sufficient clinical data for rare VUS, routine inclusion of our methods will dramatically increase the rate of VUS classification. Moreover, our calibration strategy provides a template for the development, validation, and calibration of reliable strategies for the diagnostic assessment of VUS in proteins associated with hereditary cancer predisposition syndromes and other genetic disorders.
References
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. 2015;15:181–194.
Tavtigian SV, Chenevix-Trench G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark Med. 2014;8:589–603.
LaDuca H, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;16:830–837.
Slavin TP, et al. Clinical application of multigene panels: challenges of next-generation counseling and cancer risk management. Front Oncol. 2015;5:208.
Susswein LR, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18:823–832.
Hermel, DJ, McKinnon, WC, Wood, ME & Greenblatt, MS Multi-gene panel testing for hereditary cancer susceptibility in a rural Familial Cancer Program. Fam Cancer 2016;163:383–390.
Goldgar DE, et al. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29:1265–1272.
Plon SE, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291.
Sijmons RH, Greenblatt MS, Genuardi M. Gene variants of unknown clinical significance in Lynch syndrome. An introduction for clinicians. Fam Cancer. 2013;12:181–187.
Thompson BA, et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer Family Registry. Hum Mutat. 2013;34:200–209.
Thompson BA, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2013;34:255–265.
Thompson BA, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46:107–115.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423.
Rasmussen LJ. et al. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat. 2012;33:1617–1625.
Peña-Diaz J, Rasmussen LJ. Approaches to diagnose DNA mismatch repair gene defects in cancer. DNA Repair (Amst). 2016;38:147–154.
Goldgar DE, et al. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–544.
Easton DF, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883.
Tavtigian SV. et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–1060.
Vallee MP, et al. Classification of missense substitutions in the BRCA genes: a database dedicated to Ex-UVs. Hum Mutat. 2012;33:22–28.
Drost M, et al. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat. 2010;31:247–253.
Drost M, et al. A rapid and cell-free assay to test the activity of lynch syndrome-associated MSH2 and MSH6 missense variants. Hum Mutat. 2012;33:488–494.
Drost M, Koppejan H, de Wind N. Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum Mutat. 2013;34:1477–1480.
Tavtigian, SV & Greenblatt, MS. R01CA164944: Classifying DNA Mismatch Repair Gene Variants of Unknown Significance. (2013).
Holmes J, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–5841.
Raevaara TE, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549.
Ollila S, et al. Pathogenicity of MSH2 missense mutations is typically associated with impaired repair capability of the mutated protein. Gastroenterology. 2006;131:1408–1417.
Working H, Hotelling H. Applications of the theory of error to the interpretation of trends. J Am Stat Assoc. 1929;24:73–85.
Lützen A, de Wind N, Georgijevic D, Nielsen FC, Rasmussen LJ. Functional analysis of HNPCC-related missense mutations in MSH2. Mutat Res. 2008;645:44–55.
R Foundation for Statistical Computing. RDCR: a language and environment for statistical computing. Vienna, Austria; 2015.
Hinrichsen I, et al. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin Cancer Res. 2013;19:2432–2441.
Medeiros F, Lindor NM, Couch FJ, Highsmith WE. The germline MLH1 K618A variant and susceptibility to Lynch syndrome-associated tumors. J Mol Diagn. 2012;14:264–273.
Moghadasi S, et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J Med Genet. 2018;55:15–20.
Rehm HL, et al. ClinGen—the Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242.
Vickers AJ. Decision analysis for the evaluation of diagnostic tests, prediction models and molecular markers. Am Stat. 2008;62:314–320.
Acknowledgements
We appreciate the assistance of John-Paul Plazzer, curator of the International Society for Gastrointestinal Hereditary Tumors (InSiGHT) Database. A.B.S., D.E.G., L.J.R., M.S.G., R.H.S., N.D.W., S.S.W., and S.V.T. are supported by US National Institutes of Health (NIH) National Cancer Institute (NCI) grant R01 CA164944. A.B.S. is supported by an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellowship (ID1061779). B.A.T. is supported by an NHMRC CJ Martin Early Career Fellowship. D.G. was supported in part by an NHMRC grant (ID1109286) G.K. is supported by Harboefonden (grant number 15292), Familien Spogárds Fond, and Fabrikant Einer Willumsens Mindelegat. K.M.B., L.P., and S.V.T. are supported by US NIH NCI grant P30 CA042014. L.J.R. is funded by Nordea-fonden and the Olav Thon Foundation. N.D.W. is funded by the Dutch Digestive Foundation grant FP 16-012.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Drost, M., Tiersma, Y., Thompson, B.A. et al. A functional assay–based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet Med 21, 1486–1496 (2019). https://doi.org/10.1038/s41436-018-0372-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0372-2
Keywords
This article is cited by
-
Large-scale preparation of fluorescence multiplex host cell reactivation (FM-HCR) reporters
Nature Protocols (2021)
-
Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework
Genome Medicine (2020)
-
Detection of DNA mismatch repair deficient crypts in random colonoscopic biopsies identifies Lynch syndrome patients
Familial Cancer (2020)
