
Abstract
Purpose
Glycogen storage disease (GSD) types VI and IX are rare diseases of variable clinical severity affecting primarily the liver. GSD VI is caused by deficient activity of hepatic glycogen phosphorylase, an enzyme encoded by the PYGL gene. GSD IX is caused by deficient activity of phosphorylase kinase (PhK), the enzyme subunits of which are encoded by various genes: ɑ (PHKA1, PHKA2), β (PHKB), ɣ (PHKG1, PHKG2), and δ (CALM1, CALM2, CALM3). Glycogen storage disease types VI and IX have a wide spectrum of clinical manifestations and often cannot be distinguished from each other, or from other liver GSDs, on clinical presentation alone. Individuals with GSDs VI and IX can present with hepatomegaly with elevated serum transaminases, ketotic hypoglycemia, hyperlipidemia, and poor growth. This guideline for the management of GSDs VI and IX was developed as an educational resource for health-care providers to facilitate prompt and accurate diagnosis and appropriate management of patients.
Methods
A national group of experts in various aspects of GSDs VI and IX met to review the limited evidence base from the scientific literature and provided their expert opinions. Consensus was developed in each area of diagnosis, treatment, and management. Evidence bases for these rare disorders are largely based on expert opinion, particularly when targeted therapeutics that have to clear the US Food and Drug Administration (FDA) remain unavailable.
Results
This management guideline specifically addresses evaluation and diagnosis across multiple organ systems involved in GSDs VI and IX. Conditions to consider in a differential diagnosis stemming from presenting features and diagnostic algorithms are discussed. Aspects of diagnostic evaluation and nutritional and medical management, including care coordination, genetic counseling, and prenatal diagnosis are addressed.
Conclusion
A guideline that will facilitate the accurate diagnosis and optimal management of patients with GSDs VI and IX was developed. This guideline will help health-care providers recognize patients with GSDs VI and IX, expedite diagnosis, and minimize adverse sequelae from delayed diagnosis and inappropriate management. It will also help identify gaps in scientific knowledge that exist today and suggest future studies.
Similar content being viewed by others


PURPOSE
This guideline is intended as an educational resource. It highlights current practices and therapeutic approaches to the diagnosis and management of the multiple complications of glycogen storage disease (GSD) types VI and IX.
GENERAL BACKGROUND
Overview
Glycogen is the main storage form of carbohydrate in humans. It is most abundant in liver and muscle but is also present in other tissues. Glycogen is a polymer made up of highly branched chains of glucose molecules. In the liver, glycogen acts as a glucose reserve for maintenance of blood glucose levels, especially in the fasting state. A low blood glucose level activates a series of enzymatic reactions that break down liver glycogen into glucose. The regulation of glycogen breakdown involves activation of adenylate cyclase by the hormones glucagon and epinephrine, which increases the cytosolic level of cAMP. The increased level of cAMP activates cAMP-dependent protein kinase which, in turn, activates phosphorylase kinase (PhK). PhK activates the next enzyme in the cascade, phosphorylase. Phosphorylase catalyzes the sequential cleavage of the terminal units from the glycogen chains, liberating glucose-1-phosphate, which is then converted to glucose-6-phosphate.1
At least three human glycogen phosphorylases exist, each of which is preferentially expressed in a different tissue; muscle, liver, and brain isoforms have been identified.1,2 GSD VI (OMIM 232700) is the result of a deficiency of liver glycogen phosphorylase, which is encoded by the PYGL (OMIM *613741) gene located on chromosome 14q21-q22.3 PYGL is the only gene known to be associated with GSD VI. Deficiency of muscle glycogen phosphorylase causes GSD V (OMIM 232600),4 also known as McArdle disease, and will not be discussed here.
Glycogen storage disease type IX, liver form, (OMIM 306000) (GSD IX) is often clinically indistinguishable from GSD VI. It results from deficiency of liver phosphorylase kinase (PhK). Isolated muscle PhK deficiency that is caused by pathogenic variants in PHKA1 and has also been known as GSD IXd, has also been described5,6,7,8,9,10,11 but will not be discussed in further detail here. PhK is a protein kinase that phosphorylates the inactive form of glycogen phosphorylase, phosphorylase b, to produce the active form, phosphorylase a. PhK is a heterotetramer composed of four copies each of α, β, γ, and δ subunits.12 The γ subunit contains the catalytic site. Its activity is regulated by the phosphorylation state of the regulatory α and β subunits, and by the δ subunit (calmodulin) via calcium levels.12 PhK has a wide tissue distribution with multiple tissue-specific isoforms generated by the expression and differential splicing of the various PhK subunit genes12 (Tables 1 and 2). The α-subunit is encoded by the PHKA1 (OMIM *311870) gene in muscle and by the PHKA2 (OMIM *300798) gene in liver. There are also muscle and liver isoforms of the γ-subunit, each also encoded by different genes: PHKG1 (OMIM *172470) in muscle and PHKG2 (OMIM *172471) in liver. There is only one gene encoding the β-subunit, PHKB (OMIM *172490), but it is differentially spliced in different tissues including muscle, liver, and brain.13,14 The δ-subunit of PhK, calmodulin, is encoded by three different genes—CALM1 (OMIM *114180), CALM2 (OMIM *114182), and CALM3 (OMIM *114183)—which are ubiquitously expressed and involved in other cellular processes as well. Pathogenic variants in the PHKA2, PHKB, and PHKG2 genes have been identified in patients with liver GSD IX.
History
Glycogen storage disease type VI (Hers disease) (OMIM 232700) (GSD VI) was reported by Henry-Gery Hers in 1959.15 Hers described three patients with hepatomegaly, mild hypoglycemia, an increased glycogen content and deficient activity of glycogen phosphorylase in the liver.
The first reported patient with liver PhK deficiency was described by Hug et al. in 1966.16 The patient was a female and the disorder was believed to be inherited in an autosomal recessive manner. Later in the 1960s, patients with X-linked inheritance of hepatic PhK deficiency were described.17 In some early publications, these patients were described as having a subtype of GSD VI, because they had low phosphorylase activity in addition to PhK deficiency.18,19 The term GSD IX, first designated by Hug et al.,20 was ultimately used to describe patients with primary PhK deficiency, regardless of the inheritance pattern.
Nomenclature
In older literature, GSD VI has sometimes been referred to as type VIII and IX, and GSD IX has been called GSD VIa and VIII.21 To standardize the nomenclature in this guideline paper, GSD VI will be used here to describe liver glycogen phosphorylase deficiency, and GSD IX will refer to PhK deficiency. PhK deficiency can be divided into two main types in which symptoms primarily affect liver or muscle. Liver PhK deficiency (liver GSD IX) can be further subclassified according to the gene involved. PHKA2-related GSD IX is caused by changes in the X-linked PHKA2 gene and was formerly known as GSD IXa and X-linked glycogenosis (XLG). PHKB-related GSD IX and PHKG2-related GSD IX are autosomal recessive conditions, formerly known as GSD IXb and GSD IXc, respectively.21 PHKB-related GSD IX is characterized by deficiency of PhK activity in muscle in addition to liver, but this subtype cannot be distinguished from other liver GSD IX subtypes based on clinical symptoms alone.21 Muscle PhK deficiency is also caused by pathogenic variants in PHKG1.7,10
Clinical history
As a result of phosphorylase or PhK deficiency, glycogenolysis is impaired in individuals with GSD VI and hepatic forms of GSD IX respectively (Fig. 1). The prominent clinical effects of impaired glycogenolysis include hepatomegaly, due to increased glycogen storage, and hypoglycemia. Unlike GSD I, gluconeogenesis is intact in individuals with GSDs VI and IX. Hypoglycemia consequently is usually less severe than in the classic severe form of GSD I, yet there is a spectrum of clinical severity and some patients have significant life threatening hypoglycemia. Ketosis is usually a prominent metabolic feature in both GSD VI and GSD IX22,23 that is partially due to increased fatty acid oxidation in compensation for low energy. Hepatomegaly is the most common presenting feature for patients with GSDs VI and IX usually found during routine health maintenance exams between 6 and 18 months of age. Growth retardation is also common. Patients are often diagnosed when hepatic transaminase elevation, hyperlipidemia, or ketotic hypoglycemia are found during laboratory evaluation for failure to thrive or during an acute illness. Sleep difficulties and overnight irritability are common. Due to the protean and nonspecific symptoms in GSDs VI and IX, they are almost certainly underdiagnosed. GSD IX has been diagnosed in adults who were being evaluated for hepatic cirrhosis. Unlike GSD I, lactic acid and uric acid concentrations are usually normal,24 although postprandial lactic acid can be elevated.25

Role of phosphorylase and phoshorylase kinase in the glycogenolysis pathway. GSD glycogen storage disease.
Clinical variability in GSD VI
Glycogen storage disease type VI (GSD VI) has variable severity and can present in infancy/early childhood with hepatomegaly, distended abdomen, and growth retardation. Rarely, hypoglycemia may manifest after prolonged fasting or during an illness. Ketotic hypoglycemia after an overnight fast may be seen in this disorder. Developmental delay, particularly for the motor milestones may occur in untreated children. Intellectual development is normal in most children. Severe and recurrent hypoglycemia, severe hepatomegaly, and postprandial lactic acidosis have been described in some cases.26,27 Although previously believed to be a benign condition, recent reports suggest that this is not the case. Liver fibrosis and hepatocellular carcinoma have been reported in patients with GSD VI.28,29 Presence of these complications suggests that long-term monitoring of hepatic status is necessary in GSD VI patients.
Clinical variability in GSD IX
Liver GSD IX can be divided into three subtypes based on the gene in which pathogenic variants occur (PHKA2, PHKB, and PHKG2). A wide spectrum of clinical severity, with respect to hepatomegaly, growth delay, ketotic hypoglycemia, and laboratory abnormalities has been recognized among patients with liver PhK deficiency.25,26,31,32 The basis of this clinical variation is not well understood, but some general correlations between the gene defect and clinical features have been made based on reported cases. In one or two reported cases, additional clinical symptoms have also been reported, including renal tubular acidosis, central nervous system abnormalities, and cardiomyopathy.26,28,30
PHKA2-related GSD IX
The most common subtype of liver PhK deficiency, accounting for about 75% of cases, is caused by pathogenic variants in the X-linked PHKA2 gene (OMIM *300798), and is also known as X-linked glycogenosis (XLG).33 While XLG was historically described as a mild or even benign condition, a wide range of clinical severity resulting from pathogenic variants in PHKA2 has emerged over recent years, even among individuals with the same pathogenic variant.31 As this is an X-linked condition, symptoms of liver PhK deficiency are more often seen in males. However, some female carriers also exhibit symptoms ranging from mild hepatomegaly to more severe manifestations based on X inactivation.34,35,36 Affected male children typically present in the first year or two of life with hepatomegaly, of varying degrees, and growth delay/deceleration. Further investigation often reveals mild to markedly elevated serum transaminases and hyperlipidemia. Ketotic hypoglycemia, if present, varies from occasional (only occurring after long fasts or during times of reduced intake when ill) to recurrent in some cases. Some patients have mild hypotonia in early childhood. Developmental delay has been reported. The clinical symptoms and laboratory abnormalities tend to improve with age. Puberty may be delayed, but normal height and complete sexual development can be eventually achieved.34,37,38 Most adults with X-linked liver PhK deficiency are reportedly asymptomatic.34 However, the natural history and long-term complications are not clearly delineated or understood at this time. Some patients have a relatively mild course with reports of asymptomatic hepatomegaly.39 However, at the other end of the spectrum, there are patients with severe recurrent hypoglycemia, requiring night-time tube feeding,30,31,40 and patients with liver cirrhosis due to PHKA2 and PHKG2-related GSD IX.41,42 Long-term follow-up studies are indicated in these patients to better understand the natural history of the disorder.
PHKG2-related GSD IX
Pathogenic variants in the PHKG2 (OMIM *172471) gene cause an autosomal recessive form of PhK deficiency. Pathogenic variants in the PHKG2 gene are associated with more severe clinical and biochemical abnormalities including increased risk for liver fibrosis and cirrhosis. About 25 cases have been described in the literature.28,43,44 Where information is available, most the cases reported show evidence of fibrosis on liver biopsy, and about 50% have evidence of cirrhosis.43,44 Liver cirrhosis can develop as early as the first few years of life.44,45 Occasional findings include bile duct proliferation, cholestasis, cirrhosis related esophageal varices, and splenomegaly.26,44,46,47,48 Several patients with PHKG2 pathogenic variants have been reported with liver adenomas,43,48 one with renal tubulopathy related to the development of rickets,40 and one with significant hypocalcemia.44 Muscle symptoms, including mild to moderate hypotonia, weakness, and amyotrophy, as well as delayed gross motor milestones have been reported in some patients.25,26,32,43,44,45,46 Patients with PHKG2 pathogenic variants have variable degrees of severity in their clinical spectrum.43 They tend to have more pronounced hypoglycemia requiring overnight feeding, very low PhK activity in liver, and highly elevated serum transaminases.25,26,43,44,45,46,47,48,49
PHKB-related GSD IX
Pathogenic variants in the PHKB (OMIM *172490) gene cause an autosomal recessive form of PhK deficiency. The clinical symptoms of fewer than 20 patients have been reported all of whom have liver involvement ranging from less severe to severe.14,25,28,40,50,51 Patients typically come to medical attention due to hepatomegaly. Hypoglycemia can be mild. Liver fibrosis was reported in one patient50 and an adenoma-like mass was described in another.28 Interventricular septal hypertrophy was found in one patient.28 The PHKB gene is widely expressed and differentially spliced in different tissues; exon 26 is muscle specific, and exon 27 is present in nonmuscle PhKB transcripts, including liver.13,52 Therefore, most pathogenic variants in PHKB are expected to cause PhK deficiency in liver and muscle. Despite this, muscle symptoms are either mild or absent, and patients with this subtype cannot be distinguished from those with PHKA2 or PHKG2 pathogenic variants on clinical basis alone.
Epidemiology
There are no good studies determining the prevalence of GSD VI or IX. There is a paucity of cases confirmed by genetic testing in the literature, and it is believed that these conditions are almost certainly underdiagnosed. Published prevalence estimates for GSD VI range from 1 in 65,000 to 1 in 1,000,000, but the best estimate is 1 in 100,000 individuals.53 The only known population at increased risk for GSD VI is the Mennonite community, which has a prevalence of 1 in 1000.53 There also appears to be an increased prevalence of GSD VI in northern Africa, but the frequency in Egypt, Libya, Tunisia, and Algeria is not known.25
GSD IX is one of the most common forms of glycogen storage disease, accounting for about 25% of cases.1 The frequency of liver PhK deficiency was estimated to be 1 in 100,000,24,46 but recent studies have suggested that PhK deficiency may be one of the most identifiable causes of hypoglycemia in males. It seems to be more common than GSD VI. GSD VI is equally prevalent in males and females, but GSD IX is more common in males due to pathogenic variants in the X-linked gene, PHKA2, which accounts for about 75% of cases.25,26,28
A common PHKA2 pathogenic variant in Dutch patients with liver GSD IX, p.Pro1205Leu, has been identified31 but it is not known whether GSD IX is more common in the Netherlands compared with other populations. Both GSDs VI and IX have often been a diagnosis of exclusion for which there are now improved approaches to diagnostic confirmation using molecular testing. Such approaches may lead to improved diagnosis and understanding of the true prevalence of these disorders.
METHODS/PROCESS
Consensus development panel
The evidence basis for diagnosis, management, and treatment of GSDs VI and IX is limited to expert opinion and small observational studies. As such, a national group of experts in (1) clinical and laboratory diagnosis; (2) treatment and management (nutritional, hepatic, musculoskeletal, and obstetrical); and (3) genetic aspects of GSDs VI and IX was assembled to review the evidence base and develop management guidelines. Following a meeting during which published material and personal experience were reviewed by the panel, experts in the various areas reviewed the literature in their areas and drafted the guidelines. The following terms were included in the search of PubMed: Hers disease, glycogen storage disease type VI, glycogen storage disease type IX, phosphorylase kinase deficiency, and glycogen phosphorylase deficiency. The participants provided conflict of interest statements and their conflicts are stated in the Disclosure section. All members of the panel reviewed and approved the final guidelines. Consensus was defined as agreement among all members of the panel. For the most part, the evidence and resulting recommendations are considered expert opinion because additional levels of evidence were not available in the literature. As is typical for descriptions of rare diseases, literature may be biased by more severe forms of disease. Penetrance data is limited to patient and family ascertainments. Penultimate drafts of these guidelines were shared with an external review group consisting of Yuan-Tsong (YT) Chen, MD, PhD, Annette Feigenbaum, MD, Nicola Longo, MD, PhD, and Saadet Mahmutoglu, MD. The working group considered their suggestions and changes were made as considered appropriate.
Target audience
These guidelines are directed at a wide range of care providers. Although care is commonly provided by metabolic disease specialists/biochemical geneticists, gastroenterologists, and endocrinologists in conjunction with a clinical nutritionist (dietitian), it is important that primary care providers and other specialists who often are involved in the care of individuals with GSDs VI and IX also can recognize the condition and provide appropriate care for these patients.
DIAGNOSIS
Differential diagnosis: GSD VI and liver GSD IX
The principal differential diagnosis for GSD VI and the liver GSD IXs includes other forms of GSD associated with hepatomegaly and hypoglycemia, especially GSD I and III (Table 3). Ketosis may be a clue to the correct diagnosis, as well as male gender and family history of males affected on the maternal side of the family in the X-linked form. Ultrasound imaging of the liver at baseline demonstrating isolated hepatomegaly, and the presence of nephromegaly with hypoglycemia, hypertriglyceridemia, hyperuricemia, and lactic acidosis suggest the diagnosis of GSD I (OMIM 232200).54 On liver biopsy, GSD I patients typically show extensive hepatic steatosis with some glycogen accumulation, whereas GSD III (OMIM 232400), VI, and IX patients all have extensive glycogen accumulation in the liver leading to swollen hepatocytes on electron microscopy. Additionally, in GSD III and IX there may be periportal liver fibrosis early on as well as extensive fibrosis and cirrhosis in the later stages of the disease.
Hepatomegaly, hypoglycemia, and hyperlipidemia are common features of GSDs I, III, VI, and IX. Some key differences help to differentiate these disorders. Patients with GSD I typically present in the first few months of life with severe fasting hypoglycemia within 3–4 hours after a feed. There is an associated lactic acidosis and hyperuricemia that is typically not seen in other GSDs. Usually, hypoglycemia is not as severe in patients with GSDs VI and IX because gluconeogenesis is intact; yet it needs to be recognized that some patients can present with severe recurrent hypoglycemia.7,31,43,45 Blood ß-hydroxybutyrate (ß-OHB) levels increase only modestly in GSD I22,55 as it is considered a hypoketotic hypoglycemic state. In contrast, hyperketonemia with fasting hypoglycemia is more common in GSDs III, VI, and IX.22 Hepatic transaminase levels (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) are significantly higher in GSD III, VI, and IX as compared with GSD I. In GSD I AST and ALT are usually modestly increased to ~100 U/L in the early stages of the disease with a tendency to normalize or be very mildly elevated later. Although elevated transaminase levels and hepatomegaly are common to many primary liver diseases and other metabolic disorders, hypoglycemia is distinctly uncommon until the development of end-stage liver disease for most disorders, including GSD IV, Anderson disease (OMIM 232500). The extent of hepatomegaly is comparable in GSDs VI and IX and both disorders may be associated with hyperketonemia after an overnight fast. Whereas patients with GSDs VI and IX were thought to be relatively mildly affected, there is an increasing understanding of the disease and there are patients being reported who are severely affected and closely resemble those with GSD III. GSD VI is an autosomal recessive condition and GSD IX has subtypes that are autosomal recessive and X-linked. Males are more likely to have GSD IX due to a PHKA2 pathogenic variant than they are to have GSD VI, but males can also be affected with GSD VI or other GSD IX subtypes. Females can be affected with either the autosomal recessive subtypes or rarely the X-linked subtype as manifesting heterozygotes.31,36 The ethnicity of the patient can also be considered because there is a Mennonite GSD VI founder variant and Dutch founder variant in PHKA2.31
GSD IV can be distinguished from GSDs VI and IX by absence of hypoglycemia and ketosis with progressive liver dysfunction leading to liver cirrhosis as well as accumulation of abnormally structured glycogen, resembling plant-like fibers of amylopectin. Patients with GSD III also have an abnormally structured glycogen, resembling limit dextrin. Fructose-1,6-bisphosphatase deficiency, a gluconeogenic disorder of fructose metabolism, mitochondrial disorders, and glycerol kinase deficiency have some features that may be confused with GSDs VI and IX (Table 3) though with less hepatomegaly.
Secondary PhK deficiency can be observed with Fanconi–Bickel syndrome (GSD XI)56 and cardiac/muscle glycogenosis caused by PRKAG2 deficiency (OMIM 261740)57 (Table 3). Secondary PhK deficiency can also be seen in some mitochondrial disorders including complex I deficiency. Because of severe hepatomegaly, other metabolic disorders such as Gaucher disease and Niemann–Pick type B disease may, initially, be confused with GSDs VI and IX. In both these storage diseases, however, there is striking splenomegaly, which is an important distinguishing feature, and hypoglycemia does not occur.
If the patient initially presents with hypoglycemia without appreciation of hepatomegaly, the differential diagnosis for hypoglycemia will also include GSD 0, fatty acid oxidation disorders, defects of gluconeogenesis such as fructose-1, 6-bisphosphatase deficiency, disorders of carbohydrate metabolism such as hereditary fructose intolerance, and endocrinopathies such as hyperinsulinemia, adrenal insufficiency, growth hormone deficiency, or other causes.
CLINICAL AND LABORATORY EVALUATION
Initial workup (see Table 4) in patients presenting with hepatomegaly and hypoglycemia include liver ultrasound, serum transaminases (AST, ALT), ɣ-glutamyl transferase (GGT), liver function tests (prothrombin time, albumin), blood glucose, lactate, uric acid, basic chemistry, creatine kinase (CK), plasma total and free carnitine, acylcarnitine profile, urinalysis, urine organic acids, cholesterol, triglycerides, and complete blood count (CBC) with manual differential white cell count. It is important to check for presence of plasma ketones as serum β-OHB during episodes of hypoglycemia because that would help separate ketotic hypoglycemia from nonketotic or hypoketotic hypoglycemia conditions. Measurement of insulin, growth hormone, cortisol, and free fatty acids during a critical sample of hypoglycemia is indicated to rule out endocrine causes, particularly when hepatomegaly is not a significant feature. A more detailed workup for individuals presenting with hypoglycemia and hepatomegaly is available58
At presentation, individuals with GSDs VI and IX typically have elevated transaminases. As a group, transaminase levels tend to be higher in patients with GSD IX compared with GSD VI, although there is a lot of variability between patients.25,28 GGT varies from normal to elevated.28 Because there is no overt muscle involvement in GSDs VI and IX, CK concentration is usually normal but a slight elevation can occur due to profound protein deficiency. Usually uric acid level and lactate are normal; occasionally, lactate can be elevated postprandially.25 If there is concern about falsely elevated lactate due to use of tourniquet during phlebotomy or difficult blood draw, the results of the basic metabolic panel may be helpful. Triglyceride and cholesterol levels are often elevated. Abdominal ultrasound typically reveals mild to marked diffuse hepatomegaly, often with increased liver echogenicity.28
Liver histology
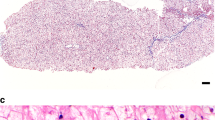
The presence of hepatomegaly often prompts a gastroenterologist to recommend a liver biopsy. However, if GSD VI or IX are suspected, a liver biopsy is not recommended to establish the diagnosis. In some cases, there is a role for liver biopsy, when no definitive diagnosis can be made noninvasively. There should be careful handling of the liver biopsy specimen to avoid loss of glycogen. Liver histology findings share features in common between GSD VI and GSD IX, and, there are distinguishing features such as presence of periportal fibrosis with thin septa in between lobules in GSD IX, often noted even in early stages of the disease. Liver parenchyma shows a mosaic of hepatocytes that are distended because of excessive glycogen accumulation in GSD VI and IX. Cell membranes are coarse and may have an undulated appearance. Scattered cytoplasmic vacuoles are present. Glycogen staining with periodic acid–Schiff (PAS) stain is diastase digestion–sensitive. The glycogen structure by electron microscopy shows excessive glycogen accumulation. The glycogen often has a frayed or burst appearance and is less compact than GSD I or III.23,59 Cytoplasmic lipid bodies are more likely to be present in hepatocytes in GSD IX. Children with GSD IX often show fibrosis of the portal tracts that may also be associated with inflammation.46 Liver cirrhosis may be variably present. It is more likely detected in individuals with PHKG2 pathogenic variants, but is also noted in some individuals with PHKA2 pathogenic variants.25,31,41,45,47 Fibrosis, but not cirrhosis, has also been reported in GSD VI.28
Biochemical analysis: glycogen content and enzyme activity
Snap frozen liver biopsies show markedly elevated glycogen content with normal structure in patients affected with GSD VI and GSD IX (glycogen content is typically 2–4 times the normal level). Glycogen structure in the liver is normal, as indicated by a normal G-1-P to glucose ratio. This distinguishes GSD VI and liver GSD IX from GSD III, which is associated with elevated glycogen content of abnormal structure (Tables 1 and 2).
DIAGNOSTIC TESTING
Background
Diagnosis by DNA analysis is preferable to liver biopsy so that patients can avoid an invasive procedure. Use of next-generation sequencing (NGS) panels, which include genes for known liver GSDs and disorders of fructose metabolism, can be helpful in making the diagnosis and distinguishing disorders with a similar presentation.58 While most clinical laboratories offer gene sequencing panels, they also include Sanger sequencing to fill in exonic sequences that are poorly covered by NGS; this should be confirmed with the individual laboratory prior to ordering testing.61,62 Exome sequencing (ES) is also being more widely used and can facilitate diagnosis.63 However, some sequences may have poor coverage, and Sanger filling is not possible. In addition, deletions/duplications can be missed on ES. Hence, the diagnosis may be missed. The identification of variants of unknown clinical significance by NGS panels or ES also pose a challenge, and follow up with histology and enzyme testing on a liver biopsy specimen may be required to confirm the diagnosis in these cases.
Enzyme assay
Glycogen phosphorylase
Hepatic glycogen phosphorylase activity can be measured in frozen liver biopsy tissue, leukocytes, and erythrocytes.27 However, phosphorylase activity can be normal in the blood cells of individuals with GSD VI3,26,65 and therefore, a normal result does not rule out the diagnosis. Although it is possible to measure the amount of active phosphorylase as well as total phosphorylase activity, it is not very reliable. It is important to note that in individuals with liver GSD IX the active phosphorylase activity and active/total phosphorylase ratio may be falsely low due to lack of activation by PhK. PhK activity should be measured in such cases to ensure that the correct diagnosis is made. Because active phosphorylase is very labile, measurement of total phosphorylase activity, which involves in vitro activation of the enzyme, is more accurate when making a diagnosis of GSD VI.
Phosphorylase b kinase
PhK activity can be measured in frozen liver biopsy, erythrocytes, leukocytes, and frozen muscle biopsy tissue. Interpretation of results of PhK activity is complex because both false positive and false negative results can occur. In a subset of patients, PhK activity in vitro is normal or even elevated when measured in erythrocytes and leukocytes, and varies from deficient to normal when measured in liver. This biochemical subtype is known as X-linked glycogenosis type 2 (XLG2).33,67 In contrast, X-linked glycogenosis type 1 (XLG1) is associated with deficient PhK activity when measured in liver and blood cells. Both XLG1 and XLG2 are caused by pathogenic variants in the X-linked PHKA2 gene and the clinical symptoms are indistinguishable. The reason in vitro PhK activity is normal or elevated in XLG2 is not yet well understood.30,33,67 Of note, elevated PhK activity in blood cells has also been associated with pathogenic variants in PHKB and could potentially result from pathogenic variants in other PhK subunit genes as well.14 False positive results can also occur, particularly because PhK is a labile enzyme that is highly sensitive to handling conditions and temperature exposure. Therefore, great care must be taken when storing and shipping diagnostic specimens. A control blood sample, drawn from an unrelated individual at the same time and location as a blood sample is drawn from a patient, is required for the blood cell PhK assay, and internal and external controls are used when measuring PhK activity in other tissues. In addition, PhK deficiency may occur secondary to a different primary metabolic defect such as GLUT2 (OMIM *138160) pathogenic variant in Fanconi–Bickel syndrome56 or PRKAG2 (OMIM *602743) pathogenic variant in isolated cardiac glycogenosis.57
DNA variant analysis
PYGL gene
The liver glycogen phosphorylase (PYGL gene, OMIM *613741) maps to chromosome 14q21-q22 and is composed of 20 coding exons. Identification of two pathogenic variants in trans in PYGL confirms a diagnosis of GSD VI. About 30 pathogenic variants have been reported throughout the PYGL gene.25,26,27 Most pathogenic variants are family-specific but there is a founder pathogenic variant, c.1620+1G>A, which is present in heterozygosity in about 3% of the Mennonite population and accounts for the high incidence (1:1000) of GSD VI in this population.53 Full gene sequencing of PYGL, as well as targeted pathogenic variant testing for the c.1620+1G>A pathogenic variant, is available on a clinical basis.
PhK subunit genes
Genetic testing for the liver form of GSD IX is complicated by the involvement of multiple large genes. To date, pathogenic variants causing liver GSD IX have been found in the X-linked PHKA2 gene, and in the PHKB and PHKG2 genes, both of which are autosomal. These genes are routinely included on GSD gene next-generation sequencing panels, which may prove more cost-effective than sequencing the genes individually.60
Pathogenic variants in the PHKA2 gene (OMIM 300798; Xp22.2-p22.1) are the most common cause of the liver form of GSD IX, accounting for about 75% of cases.25,35 If individual gene sequencing rather than panel approach is being taken, for a male patient, sequencing of the PHKA2 gene is recommended first, unless there is a clear indication of autosomal recessive inheritance.25 It should be noted that female carriers of a PHKA2 pathogenic variant can have symptoms.28,36 Therefore, presence of an affected female in the family does not rule out the possibility of X-linked inheritance. PHKA2 is composed of 33 exons. About 80 pathogenic variants have been reported throughout the gene (Human Gene Mutation Database) though many more have likely been identified by diagnostic laboratories. Most of these pathogenic variants are private, but a few have been identified in multiple patients.31,33 One of them, p.Pro1205Leu, is common in Dutch patients with GSD IX31,51 and has also been found in two unrelated Japanese families.68
Sequencing of autosomal genes PHKG2 and PHKB is recommended for males for whom no PHKA2 pathogenic variant has been found, females, and patients with family history consistent with autosomal recessive inheritance pattern. The choice of gene to sequence first may be influenced by clinical presentation; sequencing of PHKG2 is recommended first in any patients with liver cirrhosis25 with the caveat that some patients with PHKA2 pathogenic variants also develop cirrhosis.
The PHKG2 (OMIM *172471) gene encodes the catalytic subunit of PhK and contains 10 exons. Fewer than 30 pathogenic variants, spread throughout the gene, have been reported. The PHKB (OMIM *172490) gene is expressed in various tissues and is alternatively spliced to create tissue-specific isoforms. PHKB is a large gene with 31 exons. About 20 pathogenic variants throughout the gene have been reported. If only one pathogenic variant is found in either the PHKG2 or PHKB gene, deletion/duplication testing is recommended because large deletions and duplications can be missed by DNA sequencing.
Laboratory diagnostic testing recommendations:
-
To avoid liver biopsy, consider DNA testing first. Use of next-generation sequencing panels is recommended because multiple genes are involved.
-
Limitations of sequencing should be recognized, and the diagnosis pursued by other methods if there is a strong clinical suspicion.
-
PhK enzyme activity can be normal or elevated in blood cells, including erythrocytes, in affected individuals. Liver biopsy may be necessary for confirmation of the diagnosis if variants of unknown significance are identified by genetic testing.
-
Marked elevation of glycogen content in liver with structurally normal glycogen is consistent with GSD VI and GSD IX; phosphorylase and PhK enzyme activity can also be measured. In rare cases, PhK activity can be normal/not clearly deficient in liver in individuals with GSD IX. Both phosphorylase and PhK are labile enzymes so samples must be handled carefully.
-
PhK enzyme activity can be secondarily reduced due to a different, primary metabolic defect such as GLUT2 pathogenic variant in Fanconi–Bickel syndrome, PRKAG2 cardiomyopathy syndrome, or mitochondrial complex 1 deficiency.
OVERVIEW OF MANAGEMENT
GSDs VI and IX diseases are multisystem disorders with primary liver manifestations. Affected individuals are best managed by a multidisciplinary team led by a physician with expertise in these disorders. This may be a metabolic disease specialist/biochemical geneticist, or endocrinologist, together with a metabolic dietitian. Other specialists who may be required to manage specific manifestations of the disease include a physical therapist, gastroenterologist, social worker, and genetic counselor.
HEPATIC MANIFESTATIONS
Patients with GSDs VI and IX routinely present with hepatomegaly in the first years of life with elevated transaminases, alkaline phosphatase, and GGT. The transaminases may be significantly elevated in some cases of GSD IX in particular. Transaminases decrease with improved metabolic control and also with increased age; values are usually normal in adults. Hepatomegaly usually normalizes by the second decade of life.28,34 In patients who develop cirrhosis, transaminases may decrease in late stages of disease as the liver becomes increasingly cirrhotic and there are fewer hepatocytes to damage.
In one study of 205 individuals with deficiency of the glycogen phosphorylase system (PhK or phosphorylase deficiency was unspecified), three died from liver adenomas and malignant tumors and two developed cirrhosis with esophageal varices.69 In a group of 21 individuals, 17 with GSD IX and 4 with GSD VI, fibrosis was reported in about half of the patients, including the first report of fibrosis in GSD VI.30 The frequency of cirrhosis in individuals with GSD IX is unknown, but is more often found in individuals with PHKG2 pathogenic variants where it can develop in early childhood.25,44,45,47,49 At this time three individuals with PHKA2 pathogenic variants and cirrhosis have been reported.41,42 While liver cirrhosis has not been reported in patients with PHKB pathogenic variants, monitoring the liver is important.
Clinical and imaging studies
Liver enzymes are typically elevated at presentation but decrease with time so could be related to improving metabolic control or to progression of liver disease. Serum transaminases, albumin and alkaline phosphatase, GGT, prothrombin time (PT) and international normalized ratio (INR) should be measured at baseline, and followed regularly at variable intervals (3–12 months) and used as markers of liver cirrhosis. Prealbumin should also be measured as a nutritional marker to see if protein intake is adequate. Because long-term natural history for this disease is not well known or understood, some cases could be lost to follow up while they are doing well, and subsequently develop liver cirrhosis. This is especially true in patients with GSD IX.
Liver ultrasound is recommended every 12–24 months for children <18 years. With advancing age, computed tomography (CT) scan or magnetic resonance imaging (MRI) using intravenous contrast should be considered to evaluate for complications of liver disease including cirrhosis and adenomas.
Liver transplantation
As liver manifestations usually improve with conservative treatment, liver transplantation is rarely needed. However, as there is a spectrum of clinical severity, with some patients having significant liver disease, and as we continue to learn more about the natural history of the disease, liver transplant may be indicated in cases with advanced liver disease.25
Hepatic:
-
Laboratory testing to include serum AST, ALT, serum albumin, GGT, PT, INR and alkaline phosphatase every 3–12 months to monitor the extent of liver damage and as an assessment of metabolic control.
-
Abdominal ultrasound every 12–24 months in children <18 years of age; abdominal CT/MRI imaging with contrast in older patients, every 1–2 years or as indicated clinically.
-
Clinical spectrum is variable, especially hepatic manifestations and disease progression.
-
The long-term natural history of GSDs VI and XI is still emerging, There are reports of liver adenomas and cirrhosis, the latter particularly more observed in GSD IX, suggesting it is not necessarily a benign disorder.
KETOSIS/HYPOGLYCEMIA
Generally, ketosis, with or without hypoglycemia, occurs mainly during conditions of increased glucose demand and utilization with limited glycogen stores due to fasting/poor intake, pregnancy, or catabolism due to vomiting, diarrhea, or infection. Ketone bodies including 3-β-hydroxybutyrate (β-OHB), acetoacetate, and acetone act as secondary fuels during periods of low glucose availability. β-hydroxybutyrate and acetoacetate are the two main ketone bodies rich in energy. β-OHB is formed in the mitochondria from reduction of acetoacetate71 and is more stable than acetoacetate allowing β-OHB to be measured in blood, while acetoacetate is more volatile and detected in urine.72 Levels of ketone bodies vary among individuals depending on age, glycogen stores and carbohydrate availability, duration of fasting, exercise intensity, and availability of other fuel substrates such as proteins, lactate, and glycerol.71,72 Because ketone body formation can occur in the setting of fasting or stress, a precise threshold for defining pathology is difficult to define. Normally, blood β-OHB concentrations are <0.3 mmol/L, hyperketonemia is defined as a blood β-OHB concentration over 1.0 mmol/L;73,74 levels in between fall into a gray zone between physiologic ketosis and pathologic ketosis. Urine ketones may not be detected by urine strips when the blood ketone level is <1 mmol/L. Ketoacidosis indicates a state of metabolic acidosis resulting in a low blood pH, usually caused by blood ketone elevation >3 mM.73,74 If blood level is ≥3 mmol/L, ketones will be detected in urine, and there is a risk of ketoacidosis. Anion gap acidosis due to hyperketonemia can occur in patients with ketotic forms of hepatic GSD during periods of metabolic decompensation due to poor intake, vomiting, or prolonged fasting. Patients who have chronic elevations of blood ketones tolerate hypoglycemic manifestations as ketone bodies cross the blood–brain barrier, providing an alternative source of energy to the brain and sparing glucose utilization.
KETOSIS IN GSDs VI AND IX
Ketosis in glycogen storage disease is a sign of altered glycogen metabolism and enhanced counterregulatory hormone production associated with inadequate production of glucose from the liver. Stimulation of counterregulatory hormones to maintain normoglycemia (including glucagon, epinephrine, and growth hormone) acts by suppression of insulin, leading to increased lipolysis and worsening ketosis.75
Fernandes and Pekaar speculated that patients with phosphorylase deficiency compensate for low glucose production by mobilizing protein substrates for gluconeogenesis.22 This depletes gluconeogenic amino acid precursors and citric acid cycle (CAC) intermediates. Although fatty acid oxidation is increased, the exhaustion of CAC intermediates (as oxaloacetate) limits channeling of acetyl-CoA produced from fatty acid oxidation into the CAC leading to accumulation of acetyl-CoA, which is converted into 3-ketobutyryl-CoA, and eventually ketone bodies. Increasing carbohydrate intake in phosphorylase deficiency was noted to favor glycolysis over gluconeogenesis, thereby replenishing oxaloacetate and CAC intermediates, which leads to suppression of ketosis.22 High protein intake may also be beneficial in this disorder by repletion of protein precursors necessary for maintaining gluconeogenesis. High protein intake is now standard of care in the treatment of GSD III, another form of ketotic hypoglycemia. The speculation of depletion of CAC intermediates in GSDs raises concerns for anaplerotic defects, raising questions regarding a possible role for anaplerotic agents as mentioned below.
Serum β-hydroxybutyrate monitoring
Chronic ketosis in GSDs VI and IX is an indication of poor metabolic control and hormonal dysregulation, which can affect growth and bone health.76 In some patients, serum β-OHB may show a rise in the blood before blood glucose drops indicating the need to treat the child before hypoglycemia occurs. Measuring serum β-OHB in this case is more sensitive because blood levels are detectable before urine ketones. Improved nutrition, with maintenance of normoglycemia, has been associated with decreased ketones and better outcomes.76 In one study of 164 children with ketotic hypoglycemia 20 individuals (12%) were noted to have GSD (4 patients had GSD 0, 2 GSD VI, 12 GSD IX ɑ, 1 GSD IX β, and 1 GSD IX ɣ). Measuring blood glucose and β-hydroxybutyrate helped during the initial assessment of the patient’s metabolic state and follow up, and with making the diagnosis.77
Monitoring blood glucose and ketones overnight (every 3–4 hours and upon waking), prior to meals and snacks, and after activity for at least 2–3 days may be helpful in discerning the diagnosis of GSD in children with recurrent hypoglycemia with/without hepatomegaly. Once the diagnosis is established, metabolic control is monitored by measuring both blood glucose and ketone levels using glucometers and ketone meters available for home use. Blood glucose and ketone levels should be measured during times of stress including illness, intense activity, periods of rapid growth, or any time at which intake of food is reduced and before and after dietary changes are made to the amount of corn starch (CS) or protein intake. A meter that reliably measures both blood glucose and ketone levels may be used. The cost and availability of accurate meters and test strips may be burdensome for some families hence it is important to individualize the need for ketone testing and to schedule it appropriately to optimize treatment outcome while preventing excessive costs.
Implications of suppressing ketone formation as part of the treatment of GSDs VI and IX
Since gluconeogenesis is intact, protein supplementation provides gluconeogenic precursors that can be used for repletion of CAC intermediates and endogenous glucose production. By providing sufficient carbohydrate and protein, there is less dependence upon fatty acid oxidation, reduced accumulation of free fatty acids, endogenous ketone production, and enhanced gluconeogenesis.
Blood glucose and ketone monitoring:
-
Monitor blood glucose and ketone level at diagnosis and after major changes in diet, corn starch, or protein dose are made. Measure overnight (every 3–4 hours and upon waking), prior to meals and snacks, and after activity, for at least 2–3 days. At diagnosis, measure serum β-OHB. Otherwise, blood glucose and ketone measurement can be done with a home monitoring device.
-
Measure blood glucose and ketones during any times of stress such as illness, intense activity, periods of rapid growth, or any time at which intake of food is reduced.
-
Monitoring recommendations should be tailored to individual patient needs, as in some cases significant ketosis is not present.
NUTRITION
The main aim of nutrition therapy in GSDs VI and IX is to prevent the primary manifestations (hypoglycemia, ketosis, and hepatomegaly) and secondary complications (short stature, delayed puberty, and cirrhosis) by improving metabolic control. A small subset of individuals with very mild or no metabolic derangements may need no nutritional intervention. For those who experience hypoglycemia or ketosis, avoidance of fasting and small frequent feedings is recommended.1,27,32 While a high protein diet that provides 2–3 g protein/kg body weight/day is considered to be helpful and generally recommended, the distribution of calories from carbohydrates, protein, and fat is still being debated. There are three ways a high protein diet may be beneficial: amino acids derived from protein can be used as precursors for gluconeogenesis, higher dietary protein intake may also serve as a direct fuel for muscles, and glycogen storage may be reduced by replacing some of the carbohydrates with protein. Proteins from animal sources have a high biological value and are a good source of the gluconeogenic amino acids. Animal foods also provide three to seven times more protein per serving than vegetarian sources and effectively meet the dietary recommendations for GSDs VI and IX. Commercially available protein supplements are helpful in meeting the protein recommendations when dietary intake is not adequate.
In contrast to GSD I, sucrose, fructose, and lactose are not prohibited, but these simple sugars should be limited to avoid excessive glycogen storage and to prevent sudden swings in levels of blood glucose and insulin. Fats should provide ~30% of total calories and should include adequate poly and monounsaturated fats to provide essential fatty acids and “heart healthy” fats. Diets rich in animal proteins tend to be higher in saturated fats and cholesterol and care should be taken to restrict these to <10% of total calories and <300 mg/day respectively.
The role of medium-chain triglycerides (MCT oil) and anaplerotic agents in inborn errors of energy metabolism is being increasingly recognized and needs to be investigated in the management of GSDs VI and IX as aforementioned. Rare case reports showed that MCT oil and a ketogenic diet have shown beneficial effects in GSD type III (reduction of transaminases, creatine phosphokinase [CPK], and stabilization and improvement of cardiac functions).77,78,79,80,81 Abnormal bone mineralization with and without osteopenia has been reported in GSDs VI and IX.81 Dietary deficiencies and chronic ketosis are speculated to be contributory factors. Regular nutritional evaluations to assess intake of calcium and vitamin D and monitoring of 25-OH vitamin D level is recommended. Because all food groups are allowed in the diet for GSDs VI and IX, recommendations for vitamin and mineral supplementation are based on individual patient’s diet and nutrient needs.
Even though CS has been introduced in some patients as early as 6 months of age, it may not be well tolerated in infants until age 12 months because the digestive enzyme amylase may not be fully functional before this age. Generally, the requirement for CS per kg body weight (BW) is less in GSD VI and IX compared with GSD I. It is recommended for GSD VI and IX to start with a small dose of CS and gradually increase the dose based on BG levels and tolerance. Children may be able to maintain normal BG levels for 4–8 hours with 1 g/kg BW CS at bedtime. Adults need less CS per kg BW compared with children due to fewer calorie requirements relative to BW and better ability to regulate oral intake. Overnight CS dose should be titrated by checking mid-night and early morning BG levels. Daytime BG should be checked between meals and after intense physical activity to determine the need for or increase in the dose of CS.
It is important to understand that both overtreating and undertreating with CS can be problematic. Giving too much CS, too much infant formula, or too large meals can result in excess glycogen storage in the liver. Overtreating with CS can also cause diarrhea, excessive weight gain, and insulin resistance. Undertreatment is of equal concern in GSDs VI and IX, and hyperketosis can occur in the setting of relatively normal glucose concentrations because gluconeogenesis and fatty acid oxidation are intact. The goal of treatment is to maintain normal blood glucose and ketone concentrations using appropriate amounts of corn starch. The blood glucose (BG) should range between 70 and 100 mg/dL, and the target range for blood ketones is 0.0–0.2 mmol/L. Levels of the latter can be higher after overnight fast, and this is physiologic. Some patients with GSDs VI and IX do not become hyperketotic and therefore, monitoring needs to be individualized based on clinical severity. The CS amount and schedule may need revision depending on BG/ketone results possibly along with an adjustment in the protein and carbohydrate content of the diet and changes in the timing of meals and snacks. The use of an extended release corn starch from waxy maize (Glycosade®) has proven to be beneficial in children over 5 years and adults to extend the time to overnight hypoglycemia. In one study,82 efficacy of the product was demonstrated in subjects with GSD 0, III, VI, and IX by prolonging the overnight fast duration.
General nutrition recommendations
Protein:
-
Diet should be high in protein and provide 2–3 g protein/kg body weight or ~20–25% of total calories.
-
Protein intake should be distributed throughout the day.
-
Protein should be consumed at each meal and snack, before bedtime, and before physical activities.
-
Animal foods provide protein of high biological value and provide more protein per serving compared with vegetarian sources.
Carbohydrate:
-
Carbohydrates should provide ~45–50% of total calories.
-
Complex carbohydrates should be consumed with each meal to provide a sustained source of glucose.
-
Corn starch (CS) ~1 g/kg body weight may be required at bedtime to prevent overnight hypoglycemia. In some situations, CS feeding maybe required mid-night and at more frequent intervals. Glycosade (extended release CS) is tolerated well in the ketotic forms of GSD. Dosing is not the same as with CS. Overtreatment with CS can be detrimental.
-
Moderate amounts of dairy and fruits are allowed in the diet.
-
Foods high in simple sugars should be consumed in limited amounts.
Fats:
-
Fats should provide ~30% of total calories.
-
Diet should include good sources of poly- and monounsaturated fatty acids.
-
Saturated fats should provide <10% of total calories.
-
Cholesterol should be restricted to <300 mg per day.
GENERAL MEDICAL CARE
All patients with GSDs VI and IX should have a primary care provider (“medical home”) specializing in pediatrics, adolescent, or internal medicine depending on the patient’s age. The primary physician should take care of the regular physical exams, immunizations, as well as any intercurrent medical problem not related to the GSD. The primary physician should be familiar with the major manifestations of GSDs VI and IX and should maintain good communication with the patient’s specialists as needed. Some patients/families find it useful to have a binder/flash drive where they can keep physician cards, insurance information, authorizations, school evaluations, and/or other important documents.
Routine immunizations should be given as recommended by the Centers for Diseases Control and Prevention (CDC) schedule (http://www.cdc.gov/vaccines/recs/schedules/dafault.htm). Other available immunizations, like seasonal influenza, hepatitis B, pneumococcal vaccine (polyvalent after 2 years of age) should be offered, as they can prevent the hypoglycemia caused by the gastrointestinal manifestations associated with the disease processes. Hepatitis C status should be monitored in patients at risk.
Patients and their health-care providers should be aware of the potential side effects of several medications. Agents that are most likely to cause hypoglycemia are insulin and insulin secretagogues (the sulfonylureas). β-blockers can mask the symptoms of hypoglycemia. In addition, patients with muscle involvement must be cautious regarding lipid lowering agents such as simvastatin and medications such as succinylcholine that can cause rhabdomyolysis. Glucagon should not be used to treat hypoglycemia due to defective glycogenolysis. Amoxicillin is an acceptable antibiotic but Augmentin, which can cause malabsorption and contains clavulanic acid with a risk of idiopathic liver disease, is not recommended. The development of liver adenomas is at this time considered rare, yet has been reported.28,43,49 Growth hormone therapy is not indicated in GSD unless growth hormone deficiency has been proven and after nutritional therapy has been optimized. Growth hormone therapy is concerning for the potential development of liver adenomas in GSD, and it may exacerbate ketone formation.
SURGERY/ANESTHESIA
A metabolic crisis may be precipitated by prolonged fasting or illness in GSD VI and IX. Febrile illness can increase glucose requirements, and gastrointestinal illness can make it difficult or impossible to tolerate frequent oral feedings. All patients with GSD VI and IX should have an emergency letter to guide physicians who may be unfamiliar with managing acute decompensations in these disorders. This letter should describe the condition, and it must state that the patient needs to be seen immediately upon arrival to the emergency department or urgent care center. When intravenous dextrose support is required, a concentration of 10% dextrose should be used at a rate that is 1–1.25 times the maintenance rate with appropriate electrolytes. The rate can be increased based on blood glucose levels. Fluids with less concentrated dextrose, i.e., 5% dextrose, could result in fluid overload at the rate required to maintain blood glucose above 70 mg/dL and prevent ketosis. Blood glucose and β-OHB concentrations should be measured upon arrival, and blood glucose should be measured hourly on intravenous fluids until it is determined they are stable and greater than 70 mg/dL. Dextrose support should be weaned over a 2–3 hour period once full enteral intake is tolerated. As getting results such as β-OHB often takes a long time, ketone blood strips that the patient uses on a routine basis for home monitoring can also be used and provide results while awaiting the results from the blood draw.
Prolonged fasting is often required in preparation for surgery. If the patient must fast for a duration that exceeds what is usually tolerated, the patient should be admitted to the hospital the night before the procedure for intravenous dextrose support at a rate and concentration to maintain blood glucose concentration above 70 mg/dL and to prevent ketosis.32 Careful perioperative monitoring is recommended given the possibility of respiratory and metabolic complications during surgery and anesthesia. Monitoring for hypoglycemia should occur during any surgical procedure and monitoring for ketosis should be considered. In cases of cirrhosis or hepatic fibrosis, anesthetic agents with known negative effects on the liver should be avoided. Postsurgery nutrition recommendations should be directed by the surgical team depending on the procedure. Once full oral intake of meals, corn starch, and protein is tolerated, intravenous dextrose support can be safely weaned over a period of 2–3 hours.
General medical care recommendations:
-
Routine immunizations should be offered, as recommended by the CDC, including hepatitis B.
-
Medical alert bracelet should be worn and emergency care letter available for emergency management of hypoglycemia.
-
Medications that can cause hypoglycemia or liver damage should be used with caution. In patients with any muscle involvement, avoidance of agents that increase risk of rhabdomyolysis or myopathy.
-
Avoid prolonged fasting, e.g., during surgery, illness.
GYNECOLOGICAL/OBSTETRICAL CARE
Polycystic ovarian syndrome (PCOS)
PCOS has been reported in patients with GSDs VI and IX.83 Thus, the clinician must be aware of and consider evaluation in female patients with symptoms of PCOS.
Pregnancy
A pregnant patient with GSD VI or IX must be vigilant in monitoring for hypoglycemia and ketosis during pregnancy. The goal is to maintain euglycemia throughout pregnancy to prevent morbidity and mortality to the fetus due to the activation of counterregulatory hormones resulting in lipolysis and ketosis. The nutrition plan for some may only require a regular healthy diet, but for others it may require frequent snacking. Increasing protein intake may be necessary to provide an alternate source for glucose via gluconeogenesis.
Ob/gyn recommendations:
-
PCOS has been reported.
-
There should be close monitoring for hypoglycemia and ketosis during pregnancy. Goal is to maintain euglycemia throughout pregnancy.
-
Increasing protein intake may be necessary to provide an alternate source for glucose via gluconeogenesis.
PHYSICAL ACTIVITY
Contact sports at any age should be avoided if there is hepatomegaly.
OSTEOPENIA/OSTEOPOROSIS
Poor metabolic control may lead to growth failure, delayed puberty, ketotic hypoglycemia associated with altered hormones, and osteopenia/osteoporosis.27 Aggressive nutritional treatment aimed at maintaining normoglycemia and suppressing ketone formation has been associated with improved liver functions, growth, and eventually bone health.27,40
A baseline dual-energy X-ray absorptiometry (DEXA) scan should be done when puberty is complete, and subsequently as clinically indicated.84
RENAL MANIFESTATIONS
Renal tubular acidosis has been reported in two individuals with liver GSD IX. For one patient, a male with a pathogenic variant in PHKA2, the proximal renal tubular acidosis improved after the initiation of corn starch therapy.26,84 The other patient, a female with pathogenic variants in PHKG2, had renal tubulopathy related to the development of rickets associated with an inappropriate parathyroid response.40 If there are concerns, tubular resorption of phosphate can be calculated.
Follow up should be based on baseline assessments and as clinically indicated.
CARDIAC MANIFESTATIONS
In a recent case series, asymptomatic left ventricular and septal hypertrophy was reported in a patient with GSD VI, and interventricular septal hypertrophy was found in a patient with GSD IX caused by pathogenic variants in PHKB.28 The authors recommended echocardiogram every 1–2 years for patients with GSD VI and GSD IX after 5 years of age. A systematic review of the literature has not revealed other individuals with GSD VI or GSD IX and cardiac problems. Further studies are needed to determine the utility of echocardiograms in these patients. At this time, the recommendation is to perform as clinically indicated. It is also important to ensure the cardiac issue is not due to causes related to a secondary decrease in PhK activity as in PRKAG2 cardiomyopathy syndrome where a spectrum of cardiac involvement is now recognized57 or unrelated to the underlying diagnosis of GSD VI or IX.
CARE COORDINATION AND SUPPORT
The Association of Glycogen Storage Disease US (http://www.agsdus.org/) is an organization that provides information and support to people with GSD and their families. The website provides descriptions of the various types of GSD. The AGSD also holds a medical conference each year for individuals with GSD and their families.
GENETIC COUNSELING, PRENATAL DIAGNOSIS AND SCREENING
Genetic counseling and coordination of care
Similar to other inborn errors of metabolism, genetic counseling should be offered to all parents of children with GSDs VI and IX and to adults affected with the condition. In counseling families with GSDs VI and IX, at least a three-generation pedigree from the proband should be obtained.
GSD VI is an autosomal recessive condition. PHKB-related liver and muscle PhK deficiency and PHKG2-related liver PhK deficiency are inherited in an autosomal recessive manner. De novo variant rates are expected to be infrequent, and parents of an affected individual are assumed to be carriers. The recurrence risk to parents who have had an affected child is 25%. DNA variant analysis is necessary for the identification of additional family members in the extended family who may be carriers.
PHKA2-related liver PhK deficiency is inherited in an X-linked manner. If the mother is a carrier, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers. Affected males pass the disease-causing pathogenic variant to all their daughters and none of their sons. If the affected male is a simplex case (i.e., a single occurrence in a family) and if the disease-causing variant cannot be detected in the leukocyte DNA of his mother, the risk to sibs is low but greater than that of the general population because of the possibility of maternal germline mosaicism.
Carrier testing for at-risk female relatives and prenatal testing for pregnancies at risk are possible if the disease-causing variant in the family has been identified. Several laboratories in the United States offer DNA diagnostic and/or prenatal diagnostic testing for GSDs VI and IX see www.genetests.org. Large deletions and duplications cannot be detected by sequence analysis; large deletions have been reported in PHKA2 and PHKB. Identification of carrier status in the general population is limited and not routinely offered; however, variant analysis to further refine the risk of having a child with GSD VI or GSD IX can be offered to those at risk (e.g., the spouse of a known carrier or spouse of an affected person).
Prenatal diagnostic testing is typically performed by variant analysis either on cultured chorionic villus samples or amniocytes, ideally of the proband’s previously identified pathogenic variant(s). When the pathogenic variants segregating in the family are known, molecular testing is the gold standard. Preimplantation genetic diagnosis (PGD) is also an option for families with GSD if the pathogenic variants have been identified. The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
Genetic counseling/prenatal diagnosis/screening recommendations:
-
Offer genetic counseling to all parents with a child with GSDs VI and IX and to all adults with GSDs VI and IX.
-
Collect detailed family history to inform whether the disease is following an autosomal recessive or X-linked pattern of inheritance as this has implications for prenatal diagnosis and counseling.
-
When causative pathogenic variants are known, molecular testing is the preferred method for prenatal diagnosis.
EMERGING ISSUES AND KNOWLEDGE GAPS
While we understand the core clinical and laboratory features of these conditions, there are still many areas that require further research, including identification of long-term natural history, understanding the full clinical spectrum and variability of these disorders, and improvements in diagnosis, monitoring, and treatment.
First, are adults with GSD VI or IX at increased risk for any specific health problems? At this time, only one long-term study of a large group of patients has been performed. It included 41 male patients with GSD IX, 31 of whom had the X-linked form based on family history.34 These patients were followed from <10 years old to adulthood. Clinical and biochemical abnormalities gradually disappeared and most adults were asymptomatic. Further long-term studies are needed to closely follow a large number of patients with pathogenic variants in different genes to look for increased risk for specific problems—for example, whether these patients might be at increased risk of developing liver cirrhosis or adenomas in adulthood. This is likely in patients who have underlying fibrosis as noted very early in patients with GSD IX. It is important to monitor long-term health as it is increasingly recognized that previously “benign” conditions do have long-term issues.
Second, while wide clinical variability is noted in patients with GSDs VI and IX, even in patients with the same pathogenic variant,26,31 we do not have a good understanding of the factors affecting the clinical expression of these disorders. Presumably, other genetic and environmental factors are involved in determining the clinical phenotype. Candidate genes that could influence phenotype include those encoding other proteins involved in glycogen metabolism or energy metabolism in general, or changes in genes affecting liver or muscle function such as HFE and SERPINA1 that cause hereditary hemochromatosis and ɑ-1-antitrypsin deficiency, respectively. Exome/genome sequencing studies may further uncover the genetic basis for clinical variability. Environmental factors that could impact phenotype include diet and liver pathogens such as hepatitis. Evaluation of these factors that could affect the phenotype of GSDs VI and IX will provide insight into the clinical variability of these disorders and guide management of patients.
Third, further work is needed to better understand the clinical features that may be associated with GSD VI and GSD IX. For example, there are reports of individuals with a confirmed diagnosis of GSD VI or liver PhK deficiency and documented developmental delay, including intellectual impairment or borderline intellectual functioning and/or speech delay26,30,40 (personal observation). While the majority of children with PhK deficiency do not have problems with cognition or speech, these observations have raised the question of whether PhK deficiency might impact on nervous system development. One possibility is the impact of hypoglycemia on the developing brain.86 Indeed, there is one report of a child with GSD VI and cognitive delay associated with hypoglycemic seizures.26 However, not all the observed patients with GSD IX and cognitive/global developmental delay had documented hypoglycemia and few manifest seizures. The vast majority of children with GSD IX do not have delayed development, even if hypoglycemia occurred. This suggests at least that hypoglycemia is unlikely to be the sole factor responsible for these issues. This is further supported by the overall normal cognitive profiles in individuals with GSD I who are at the highest risk for significant hypoglycemia. Of note, PhK is expressed in the brain,87 and an in silico study showed expressed sequence tags for all the gene subunits in the brain tissue.88 Therefore, it is possible that PhK may have a brain-specific function that is impacted by specific pathogenic variants. At the current time, there is insufficient evidence to conclude whether cognitive impairment might be part of the clinical spectrum seen in patients with PhK deficiency. Further work is needed to determine whether learning difficulties are, indeed, more common in patients with GSD IX compared with the general population and if so, the molecular basis behind this observation. Importantly, any individual with PhK deficiency who presents with developmental delay should be offered comprehensive evaluation for developmental delay, including fragile X DNA and chromosome analyses.88 Future studies of these patients might include exome/genome sequencing.
Fourth, there is suggestion that specific biomarkers, including urine glucose tetrasaccharide (Glc4), urine organic acids, and serum biotinidase activity could be helpful in the diagnosis and monitoring of patients with hepatic GSDs, such as GSDs VI and IX. For example, urine Glc4 is a limit dextrin of glycogen that is normally excreted in urine and is elevated in several types of GSD including II, III, VI, and IX31,35,90,91 as well as other conditions. An association between clinical condition and urine Glc4 level in patients with PHKA2 pathogenic variants has been reported.31,35 Patients with GSD IX occasionally have urine organic acid abnormalities, including elevation of 3-methylglutaconic acid and other markers of mitochondrial dysfunction.31,43 Elevation of 3-methylglutaconic acid has also been reported in GSD I and may be related to abnormal fatty acid oxidation or mitochondrial dysfunction.92 No systematic study has been done to determine whether urine organic acid abnormalities correlate with clinical status. Biotinidase activity is elevated in the serum of patients with hepatic GSDs including GSDs VI and IX93 although a recent study questioned the utility of biotinidase activity as a biomarker for these disorders due to inter- and intraindividual variability.94 Further studies are needed to evaluate these biomarkers, and potentially others, and to determine whether they are useful for diagnosing and monitoring patients with GSDs VI and IX.
In treatment for GSDs VI and IX, the benefits of a high protein diet, corn starch therapy and avoidance of long periods of fasting are well understood. But the role of anaplerotic agents as triheptanoin or alternative sources of energy such as MCT oil in preventing hypoglycemia and preventing endogenous ketosis needs to be investigated through controlled trials. Furthermore, as with other liver GSDs, including GSD I and III, long-term hepatic and other complications are emerging despite good metabolic control. There is a need for more definitive therapies for these conditions. Nutrition has altered the natural history to some extent, yet there are several complications that are being uncovered.
In summary, our knowledge of GSDs VI and IX continues to evolve and improve. However, there are still many unknowns that pose challenges when counseling and caring for families impacted by these disorders. A few of these areas have been highlighted here but there is much more to be learned. Continued research, including development of registries, could help to fill these knowledge gaps and lay the basis for future research studies.
References
Kishnani PS, Koeberl D, ChenYT. Glycogen storage diseases. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Mitchell G,Scriver’s online metabolic & molecular bases of inherited disease. New York: McGraw-Hill; 2016.
Newgard CB, Hwang PK, Fletterick RJ. The family of glycogen phosphorylases: structure and function. Crit Rev Biochem Mol Biol. 1989;24:69–99.
Burwinkel B, Bakker H, Herschkovitz E, Moses S, Shin Y, Kilimann M. Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI (Hers disease). Am J Hum Genet. 1998;62:785–791.
DiMauro S, Andreu AL, Bruno C, Hadjigeorgiou GM. Myophosphorylase deficiency (glycogenosis type V; McArdle disease). Curr Mol Med. 2002;2:189–196.
Wehner M, Clemens PR, Engel AG, Kilimann MW. Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet. 1994;3:1983–1987.
Bruno C, Manfredi G, Andreu AL, Shanske S, Krishna S, Ilse WK, et al. A splice junction mutation in the alpha(M) gene of phosphorylase kinase in a patient with myopathy. Biochem Biophys Res Commun. 1998;249:648–651.
Burwinkel B, Hu B, Schroers A, Clemens PR, Moses SW, Shin YS, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003;11:516–526.
Wuyts W, Reyniers E, Ceuterick C, Storm K, de Barsy T, Martin JJ. Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. Am J Med Genet A. 2005;133A:82–84.
Ørngreen MC, Schelhaas HJ, Jeppesen TD, Akman HO, Wevers RA, Andersen ST, et al. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurol. 2008;70:1876–1882.
Echaniz-Laguna A, Akman HO, Mohr M, Tranchant C, Talmant-Verbist V, Rolland MO, Dimauro S. Muscle phosphorylase b kinase deficiency revisited. Neuromuscul Disord. 2010;20:125–127.
Preisler N, Orngreen MC, Echaniz-Laguna A, Laforet P, Lonsdorfer-Wolf E, Doutreleau S, et al. Muscle phosphorylase kinase deficiency: a neutral metabolic variant or a disease? Neurol. 2012;78:265–268.
Brushia RJ, Walsh DA. Phophorylase kinase: the complexity of its regulation is reflected in the complexity of its structure. Front Biosci. 1999;4:D618–41.
Wüllrich-Schmoll A, Kilimann MW. Structure of the human gene encoding the phosphorylase kinase beta subunit (PHKB). Eur J Biochem. 1996;238:374–380.
Burwinkel B, Moses SW, Kilimann MW. Phosphorylase-kinase-deficient liver glycogenosis with an unusual biochemical phenotype in blood cells associated with a missense mutation in the beta subunit gene (PHKB). Hum Genet. 1997;101:170–174.
Hers HG. Enzymatic studies of hepatic fragments: application to the classification of glycogenoses. Rev Int Hepatol. 1959;9:35–55.
Hug G, Schubert WK, Chuck G. Phosphorylase kinase of the liver: deficiency in a girl with increased hepatic glycogen. Science. 1966;153:1534–1535.
Huijing F, Fernandes J. X-chromosomal inheritance of liver glycogenosis with phosphorylase kinase deficiency. Am J Hum Genet. 1969;21:275–284.
Huijing F. Phosphorylase kinase in leucocytes of normal subjects and of patients with glycogen storage disease. Biochim Biophys Acta. 1967;148:601.
Huijing F. Enzymes of glycogen metabolism in leucocytes, in relation to glycogen storage disease. In: Whelan WJControl of glycogen metabolism. New York: Academic Press; 1968. p. 115.
Hug G, Garancis JC, Schubert WK, Kaplan S. Glycogen storage disease types II, III, VIII, and IX. Am J Dis Child. 1966;3:457–474.
Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007;13:2541–2553.
Fernandes J, Pikaar NA. Ketosis in hepatic glycogenosis. Arch Dis Child. 1972;47:41–46.
Tuchman M, Brown BI, Burke BA, Ulstrom RA. Clinical and laboratory observations in a child with hepatic phosphorylase kinase deficiency. Metabolism. 1986;35:627–633.
Wolfsdorf JI, Holm LA, Weinstein DA. Glycogen storage diseases: phenotypic, genetic, and biochemical characteristics. Rev Endocr Metab Disord. 2003;4:95–102.
Davit-Spraul A, Piraud M, Dobbelaere D, Valayannopoulos V, Labrune P, Habes D, et al. Liver glycogen storage diseases due to phosphorylase system deficiencies: diagnosis thanks to non-invasive blood enzymatic and molecular studies. Mol Genet Metab. 2011;104:137–143.
Beauchamp NJ, Taybert J, Champion MP, Layet V, Heinz-Erian P, Dalton A, et al. High frequency of missense mutations in glycogen storage disease type VI. J Inherit Metab Disord. 2007;30:722–734.
Dagli A and Weinstein DA. Glycogen storage disease type VI. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle, WA: University of Washington–Seattle; 1993–2018. https://www.ncbi.nlm.nih.gov/books/NBK5941/.
Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, et al. The natural history of glycogen storage disease types VI and IX: long-term outcome from the largest metabolic center in Canada. Mol Genet Metab. 2014;113:171–176.
Manzia TM, Angelico R, Toti L, et al. Glycogen storage disease type Ia and VI associated with hepatocellular carcinoma: two case reports. Transplant Proc. 2011;43:1181–1183.
Burwinkel B, Amat L, Gray RG, Matsuo N, Muroya K, Narisawa K, et al. Variability of biochemical and clinical phenotype in X-linked liver glycogenosis with mutations in the phosphorylase kinase PHKA2gene. Hum Genet. 1998;102:423–429.
Achouitar S, Goldstein JL, Mohamed M, Austin S, Boyette K, Blanpain FM, et al. Common mutation in the PHKA2 gene with variable phenotype in patients with liver phosphorylase b kinase deficiency. Mol Genet Metab. 2011;104:691–694.
Herbert M, Goldstein JL, Rehder C, Austin S, Kishnani PS, and Bali DS. Phosphorylase kinase deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle, WA: University of Washington–Seattle; 1993–2018. http://www.ncbi.nlm.nih.gov/books/NBK55061.
Hendrickx J, Lee P, Keating JP, Carton D, SArdharwalla IB, Tuchman M, et al. Complete genomic structure and mutational spectrum of PHKA2 in patients with X-linked liver glycogenosis type I and II. Am J Hum Genet. 1999;64:1541–1549.
Willems PJ, Gerver WJ, Berger R, Fernandes J. The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients. Eur J Pediatr. 1990;149:268–271.
Morava E, Wortmann SB, van Essen HZ, Liebrand van Sambeek R, Wevers R, van Diggelen OP. Biochemical characteristics and increased tetraglucoside excretion in patients with phosphorylase kinase deficiency. J Inherit Metab Dis. 2005;28:703–706.
Cho SY, Lam CW, Tong SF, Siu WK. X-linked glycogen storage disease IXa manifested in a female carrier due to skewed X chromosome inactivation. Clin Chim Acta. 2013;426:75–78.
Dunger DB, Leonard JV, Preece MA. Patterns of growth in the hepatic glycogenoses. Arch Dis Child. 1984;59:657–660.
Schippers HM, Smit GP, Rake JP, Visser G. Characteristic growth pattern in male X-linked phosphorylase-b kinase deficiency (GSD IX). J Inherit Metab Dis. 2003;26:43–47.
Kim JA, Kim JH, Lee BH, Kim GH, Shin YS, Yoo HW, et al. Clinical, biochemical, and genetic characterization of glycogen storage type IX in a child with asymptomatic hepatomegaly. Pediatr Gastroenterol Hepatol Nutr. 2015;18:138–143.
Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, et al. Glycogen storage disease type IX: High variability in clinical phenotype. Mol Genet Metab. 2007;92:88–99.
Johnson AO, Goldstein JL, Bali D. Glycogen storage disease type IX: novel PHKA2 missense mutation and cirrhosis. J Pediatr Gastroenterol Nutr. 2012;55:90–92.
Tsilianidis LA, Fiske LM, Siegel S, Lumpkin C, Hoyt K, Wasserstein M, et al. Aggressive therapy improves cirrhosis in glycogen storage disease type IX. Mol Genet Metab. 2013;109:179–182.
Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, et al. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol Genet Metab. 2014;111:309–313.
Albash B, Imtiaz F, Al-Zaidan H, Al-Manea H, Banemai M, Allam R, et al. Novel PHKG2 mutation causing GSD IX with prominent liver disease:report of three cases and review of literature. Eur J Pediatr. 2014;173:647–653.
Burwinkel B, Tanner MS, Kilimann MW. Phosphorylase kinase deficient liver glycogenosis: progression to cirrhosis in infancy associated with PHKG2 mutations (H144Y and L225R). J Med Genet. 2000;37:376–377.
Maichele AJ, Burwinkel B, Maire I, Søvik O, Kilimann MW. Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet. 1996;14:337–340.
van Beurden EA, de Graaf M, Wendel U, Gitzelmann R, Berger R, van den Berg IE. Autosomal recessive liver phosphorylase kinase deficiency caused by a novel splice-site mutation in the gene encoding the liver gamma subunit (PHKG2). Biochem Biophys Res Commun. 1997;236:544–548.
Burwinkel B, Rootwelt T, Kvittingen EA, Chakraborty PK, Kilimann MW. Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr Res. 2003;54:834–839.
Burwinkel B, Shiomi S, Al Zaben A, Kilimann MW. Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum Mol Genet. 1998;7:149–154.
Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS, et al. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum Mol Genet. 1997;6:1109–1115.
van den Berg, van Beurden EA, Malingré HE, van Amstel HK, Poll-The BT, Smeitink JA, et al. X-linked liver phosphorylase kinase deficiency is associated with mutations in the human liver phosphorylase kinase alpha subunit. Am J Hum Genet. 1995;56:381–387.
Harmann B, Zander NF, Kilimann MW. Isoform diversity of phosphorylase kinase alpha and beta subunits generated by alternative RNA splicing. J Biol Chem. 1991;266:15631–15637.
Chang S, Rosenberg M, Holmes M, Francomano CA, Biesecker LG. Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI. Hum Mol Genet. 1998;7:865–870.
Lee P, Mather S, Owens C, Leonard J, Dicks-Mireaux C. Hepatic ultrasound findings in the glycogen storage disease. Br J Radiol. 1994;67:1062–1066.
Binkiewicz A, Senior B. Decreased ketogenesis in von Gierke’s disease (type 1 glycogenosis). J Pediatr. 1973;83:973–978.
Burwinkel B, Sanjad SA, Al-Sabban E, Al-Abbad A, Kilimann MW. A mutation in GLUT2, not in phosphorylase kinase subunits, in hepato-renal glycogenosis with Fanconi syndrome and low phosphorylase kinase activity. Hum Genet. 1999;105:240–243.
Burwinkel B, Scott JW, Bührer C, van Landeghem FK, Cox GF, Wilson CJ, et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet. 2005;76:1034–1049.
Saudubray J-M, Charpentier C. Clinical phenotypes: diagnosis/algorithms. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Mitchell GScriver’s online metabolic & molecular bases of inherited disease. New York: McGraw-Hill; 2016.
McAdams A, Hug G, Bove K. Glycogen storage disease, types I to X: criteria for morphologic diagnosis. Hum Pathol. 1974;5:463.
Wang J, Cui H, Lee NC, Hwu WL, Chien YH, Craigen WJ, et al. Clinical application of massively parallel sequencing in the molecular diagnosis of glycogen storage diseases of genetically heterogeneous origin. Genet Med. 2013;15:106–114.
Sulonen AM, Ellonen P, Almusa H, Lepistö M, Eldfors S, Hannula S, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94.
Samorodnitsky E, DattSa J, Jewell BM, Hagopian R, Miya J, Wing MR, et al. Comparison of custom capture for targeted next-generation DNA sequencing. J Mol Diagn. 2015;17:64–75.
Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46.
ACMG Board of Directors. Points to consider for informed consent for genome/exome sequencing. Genet Med. 2013;9:748–749.
Tang N, Hui J, Young E, Worthington V, To K, Cheung K, Chi-Kong L. et al. A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity. Mol Genet Metab. 2003;79:142–145.
Hendrickx J, Dams E, Coucke P, Lee P, Fernandes J, Willems PJ. X-linked liver glycogenosis type II (XLG II) is caused by mutations in PHKA2, the gene encoding the liver alpha subunit of phosphorylase kinase. Hum Mol Genet. 1996;5:649–652.
Carriere C, Mornon JP, Venien-Bryan C, Boisset N, Callebaut I. Calcineurin B-like domains in the large regulatory alpha/beta subunits of phosphorylase kinase. Proteins. 2008;99:1597–1606.
Hirono H, Shoji Y, Takahashi T, Sato W, Takeda E, Nishijo T, et al. Mutational analyses in four Japanese families with X-linked liver phosphorylase kinase deficiency type 1. J Inherit Metab Dis. 1998;21:846–852.
Hers HG, Glycogen storage diseases. Metabolic basis of inherited disease, 425–452. McGraw-Hill, New York (1989).
Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–426.
Sprague JE, Arbeláez AM. Glucose counterregulatory responses to hypoglycemia. Pediatr Endocrinol Rev. 2011;9:463–475.
Clarke W, Jones T, Rewers A, Dunger D, Klingensmith GJ. ISPAD clinical practice consensus guidelines 2006–2007: assessment and management of hypoglycemia in children and adolescents with diabetes. Pediatr Diabetes. 2008;9:165–174.
Mitchell GA, Kassovska-Bratinova S, Boukaftane Y, et al. Medical aspects of ketone body metabolism. Clin Invest Med. 1995;18:193–216.
Stralfors P, Olsson H, Belfrage P. Hormone-sensitive lipase. In: Boyer P, Krass E, eds. The enzymes. New York: Academic Press; 1987. p. 144–177.
Derks TG,J, van Rijn M. Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. J Inherit Metab Dis. 2015;38:537–543.
Brown LM, Corrado MM, van der Ende RM, Derks TGJ, Chen MA, Siegel S, et al. Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children. J Inherit Metab Dis. 2015;38:489–493.
Valayannopoulos V, Bajolle F, Arnoux J-B, Dubois S, et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III with D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr Res. 2011;70:638–641.
El-Gharbawy AH, Arnold GL, Perrott-Taylor N, Hughley T, Long K, Vockley J, et al. Optimizing metabolic control of glycogen storage disease type 3 (Gsd3): potential role for medium chain triglycerides (MCT). Mol Genet Metab. 2014;111:284–285.
Brambilla A, Mannarino S, Pretese R, Gasperini S, Galimberti C, Parini R. Improvement of cardiomyopathy after high-fat diet in two siblings with glycogen storage disease type III. JIMD Rep. 2014;17:91–95.
Miller JH, Stanley P, Gates GF. Radiography of glycogen storage diseases. Am J Roentgenol. 1979;132:379–387.
Heller S, Worona L, Consuelo A. Nutritional therapy for glycogen storage diseases. J Pediatr Gastroenterol Nutr. 2008;47 suppl 1:S15–21.
Ross KM, Brown LM, Corrado MM, Chengsupanamit T, Curry LM, Ferrecchia IA, et al. Safety and efficacy of long-term use of extended release corn starch therapy for GSD types 0, III, VI, and IX. J Nutr Ther. 2015;4:1–6.
Lee PJ, Leonard JV. The hepatic glycogen storage diseases--problems beyond childhood. J Inherit Metab Dis. 1995;18:462–472.
Nagai T, Matsuo N, Tsuchiya Y, Cho H, Hasegawa Y, Igarashi Y. Proximal renal tubular acidosis associated with glycogen storage disease, type 9. Acta Paediatr Scand. 1988;77:460–463.
Ukechuku GO, Shoemaker LR, Dambska M, Brown LM, Mathew J, Weinstein DA Tight metabolic control plus ACE inhibitor therapy improves GSD I nephropathy. J Inherit Metab Dis 2017;40:703–708.
Melis D, Parenti G, Della Casa R, Sibilio M, Romano A, Di Salle F, et al. Brain damage in glycogen storage disease type I. J Pediatr. 2004;144:637–642.
Psarra AM, Sotiroudis TG. Subcellular distribution of phosphorylase kinase in rat brain. Association of the enzyme with mitochondria and membranes. Int J Biochem Cell Biol. 1996;28:29–42.
Winchester JS, Rouchka EC, Rowland NS, Rice NA. In silico characterization of phosphorylase kinase: evidence for an alternate intronic polyadenylation site in PHKG1. Mol Genet Metab. 2007;92:234–242.
Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: genetic and metabolic testing on children with global developmental delay. Neurol. 2011;77:1629–1635.
Oberholzer K, Sewell AC. Unique oligosaccharide (apparently glucotetrasaccharide) in urine of patients with glycogen storage diseases. Clin Chem. 1990;36:1381.
An Y, Young SP, Hillman SL, Van Hove JL, Chen YT, Millington DS. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem. 2000;287:136–143.
Law LK, Tang NL, Hui J, Lam CW, Fok TF. 3-methyglutaconic aciduria in a Chinese patient with glycogen storage disease Ib. J Inherit Metab Dis. 2003;26:705–709.
Paesold-Burda P, Baumgartner MR, Santer R, Bosshard NU, Steinmann B. Elevated serum biotinidase activity in hepatic glycogen storage disorders—a convenient biomarker. J Inherit Metab Dis. 2007;30:896–902.
Angaroni CJ, Giner-Ayala AN, Hill LP, Guelbert NB, Paschini-Capra AE, Dodelson de Kremer R. Evaluation of the biotinidase activity in hepatic glycogen storage disease patients. Undescribed genetic finding associated with atypical enzymatic behavior: an outlook. J Inherit Metab Dis. 2010;33 suppl 22:S289–94.
Acknowledgements
The authors thank Yuan-Tsong (YT) Chen, Annette Feigenbaum, Nicola Longo, and Saadet Mahmutoglu for their critical comments and suggestions to this guidelines document. We also thank Mrudu Herbert for her help with editing this guideline and help with correcting molecular pathogenic variants data for GSD I.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Disclaimer
This practice resource is designed primarily as an educational resource for medical geneticists and other clinicians to help them provide quality medical services. Adherence to this practice resource is completely voluntary and does not necessarily assure a successful medical outcome. This practice resource should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinician should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen.
Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this practice resource. Clinicians also are advised to take notice of the date this practice resource was adopted, and to consider other medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this clinical practice resource on 27 August 2018.
Rights and permissions
About this article

Cite this article
Kishnani, P.S., Goldstein, J., Austin, S.L. et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 21, 772–789 (2019). https://doi.org/10.1038/s41436-018-0364-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0364-2
Keywords
This article is cited by
-
Report of an Iranian child with chronic abdominal pain and constipation diagnosed as glycogen storage disease type IX: a case report
Journal of Medical Case Reports (2024)
-
Endocrine involvement in hepatic glycogen storage diseases: pathophysiology and implications for care
Reviews in Endocrine and Metabolic Disorders (2024)
-
Clinical and genetic spectrum of GSD type 6 in Korea
Orphanet Journal of Rare Diseases (2023)
-
Glycogen storage diseases
Nature Reviews Disease Primers (2023)
-
Dietary Management of Metabolic Liver Disease
Current Hepatology Reports (2023)
