
Abstract
Purpose
Autosomal dominant polycystic kidney disease (ADPKD) is a common adult-onset monogenic disorder, with prevalence of 1/1000. Population databases including ExAC have improved pathogenic variant prioritization in many diseases. Due to pseudogene homology of PKD1, the predominant ADPKD disease gene, and the variable disease severity and age of onset, we aimed to investigate the utility of ExAC for variant assessment in ADPKD.
Methods
We assessed coverage and variant quality in the ExAC cohort and combined allele frequency and age data from the ExAC database (n = 60,706) with curated variants from 2000 ADPKD pedigrees (ADPKD Mutation Database).
Results
Seventy-six percent of PKD1 and PKD2 were sequenced adequately for variant discovery and variant quality was high in ExAC. In ExAC, we identified 25 truncating and 393 previously reported disease-causing variants in PKD1 and PKD2, 6.9-fold higher than expected. Fifty-four different variants, previously classified as disease-causing, were observed in ≥5 participants in ExAC.
Conclusion
Our study demonstrates that many previously implicated disease-causing variants are too common, challenging their pathogenicity, or penetrance. The presence of protein-truncating variants in older participants in ExAC demonstrates the complexity of variant classification and highlights need for further study of prevalence and penetrance of this common monogenic disease.
Similar content being viewed by others

Introduction
Autosomal dominant polycystic kidney disease (ADPKD), with a reported prevalence of 1 in 1000, is a common, adult-onset monogenic disease and the most common genetic cause of kidney failure.1 The disease is predominantly caused by pathogenic variants in PKD1 or PKD2. The utility of genetic testing in ADPKD is increasing as sequencing technology advances and as evidence accumulates regarding the value of a genetic diagnosis in estimating disease progression.2,3,4 In addition, genetic diagnosis provides families with valuable information regarding recurrence risk and access to techniques such as preimplantation genetic diagnosis. As therapeutic options advance, genetic diagnosis can also be used to stratify patients’ suitability for treatments.2
However, genetic sequencing in ADPKD has not been straightforward, largely due to six pseudogenes that share 97% sequence similarity to PKD1 and therefore challenge standard sequencing techniques.5 In addition, interpreting variants identified in patients with APDKD is challenging. The majority of ADPKD patients have protein-truncating variants that are considered to be disease-causing (approximately 60% of PKD1 mediated disease and 90% of PKD2 mediated disease).1 Some have missense variants that are predicted to cause their disease; however, the interpretation of these variants currently relies heavily on in silico prediction models, because functional studies to assess pathogenicity of individual variants are not readily accessible, particularly to diagnostic laboratories.6 Previous reports of pathogenicity have limited utility given most (up to 80%) have disease-causing variants unique to their family.7
Variant analysis in ADPKD is also challenged by significant variability in disease severity among patients, ranging from increased number of kidney cysts to end stage renal failure (ESRF). The increasing use of genetic diagnostics in ADPKD reinforces the necessity for reliable pathways to assess the likelihood of identified variants causing clinically significant disease, such as impaired kidney function, rather than purely predicting the development of increased number of renal cysts.
As sequencing technology has evolved, there has been re-evaluation of diagnostic guidelines for interpreting the pathogenicity of identified variants.8 In addition, large-scale population databases have become widely available and can assist in the assessment of genetic variants in rare diseases.9 There is evidence in several diseases where these databases have improved the interpretation of variants in groups of patients with monogenic disease and allowed better prediction of which variants are likely to cause clinically significant disease.10,11 The most widely used large-scale reference data set is from the Exome Aggregation Consortium (ExAC) consisting of exome sequencing data from unrelated individuals of diverse ancestries.9 The PKD Database (PKDB) contains sequencing data from over 2000 pedigrees of patients with an ADPKD phenotype.12
Here we assessed the prevalence of disease-causing variants in a population data set, by combining curated variants from over 2000 pedigrees from the PKDB12 to 60,706 individuals from the ExAC population database.9 This work extends previous studies using population databases by focusing on an adult-onset disease with variable disease severity and investigating a primary disease gene with strong pseudogene homology that has warranted detailed assessment of the quality of the data within the ExAC exome data set.13
Materials and methods
Variant data was downloaded from the ExAC database in VCF format (http://exac.broadinstitute.org; version 0.3) from 60,706 individuals, and annotated by VEP v79 (ref. 14). Only variants annotated as “PASS” by the Variant Quality Score Recalibration (VQSR) quality filter, and affecting specific PKD1 (ENST00000262304) and PKD2 (ENST00000237596) isoforms were used for analysis. All variants reported were included in analysis, including variants in the coding region, untranslated regions, and flanking introns. Protein-truncating variants (i.e., gain of stop codon, frameshift insertions or deletions, and essential splice site variants) were flagged. The reads supporting all protein-truncating variants were manually inspected for evidence of read misalignment using the ExAC browser’s interactive IGV.js browser. Two predicted nonsense variants (p.Glu4131Ter, p.Glu1295Ter) were removed because they were immediately adjacent to other nucleotide substitutions, in the same patients, on the same haplotype, which together results in a missense change. Also, one frameshift variant (p.Leu3446ArgfsTer27) was removed because it showed evidence for read misalignment. The average mapping quality was assessed for all variants directly from the ExAC VCF file. Ages for all participants with a PKD1 or PKD2 variant were obtained from ExAC. Age data were not readily available for the recently expanded gnomAD cohort, thus here we restricted our analysis to the n = 60,706 ExAC cohort.
Variant data from the PKDB (PKDB.mayo.edu; accessed 20 May 2016) were converted to VCF format (CHR; POS; REF; ALT) for cross-comparison with variants from ExAC.
Given the known sequence homology between PKD1 and the six PKD1 pseudogenes, we aimed to identify regions of PKD1 that would be most susceptible to pseudogene read misalignment with short-read exome sequencing. False positive variants could occur if reads from a pseudogene are incorrectly aligned to PKD1. To detect regions of PKD1 that are at increased risk for false positive calls, a list of regions of the PKD1 gene that are similar in sequence to a pseudogene sequence was compiled (noting that reads that are identical to two sites of the genome are discarded from analysis, mapping quality [MQ] = 0). Regions of the PKD1 gene that are almost identical in sequence to a pseudogene are most vulnerable to misaligned reads and subsequently false positive calls (Supplementary Figure 1). A list of regions in PKD1 that are most susceptible to false positive calls due to pseudogene homology was generated. A BED file for the pseudogene exons was made using coordinates from RefSeq and ENSEMBL (Supplementary Table 1; Supplementary Figure 1). The sequence of each pseudogene exon was obtained from the GRCh37 reference genome and the BED files in FASTA format, and converted to FASTQ format using bedtools,15 setting all bases to quality of 30. To direct all pseudogene exons to align to PKD1 in the subsequent step, we created a custom reference genome, by masking the PKD1 pseudogenes from the hs37d5 reference genome using bedtools (Supplementary Figure 1). FASTQ files from each pseudogene were aligned to the custom reference genome, using BWA MEM16 (Supplementary Figure 1). Variants were identified using samtools and bcftools,17 in VCF format. This VCF file represents a set of all possible false positive variants that could be attributed to pseudogene read misalignment. The generated list of variants was cross-referenced with variants in ExAC.
We estimated the maximum credible population allele frequency and allelic count for variants in ExAC, using the frequency calculator established by Whiffin et al.18 under the following assumptions: a disease prevalence of 1/1000, allelic heterogeneity of 1%, genetic heterogeneity of 100%, a penetrance of 100%, a reference population size of 121,412 alleles, and statistical confidence of 0.999.
Results
Assessing data quality in ExAC for PKD1 and PKD2
Given PKD1 shares >97% sequence homology with six pseudogenes and that ExAC is composed of exome sequencing data from short-read sequencing (paired end 75 bp), we carefully examined data quality.5,9 To investigate the potential for false positive variants resulting from read misalignment, we assessed average mapping quality (MQ; i.e., the likelihood that the reads are aligned to the correct section of the genome) of each variant reported in ExAC (Supplementary Figure 2A). As expected the MQ was lower across exons 1–33, which share homology with pseudogenes; however, all variants had average MQ >30, indicating a 1:1000 chance that reads were mapped incorrectly, suggesting that most variants are likely to be real.
We next assessed whether any variants observed in PKD1 could have been from PKD1 pseudogene read misalignment. By aligning the pseudogene sequences to PKD1 (Supplementary Figure 1), we identified a set of 1028 variants that would be consistent with pseudogene read misalignment, if observed in an individual. This file serves as a useful control for interpreting patient data (Supplementary Table 2). By comparing all variants in PKD1 in ExAC with the potential false positives, only 64 (1.34%) variants matched the pseudogene sequence and none of these were protein-truncating variants. PKD2 does not have any pseudogenes and we previously demonstrated that reads align well to PKD2 (ref. 3).
We investigated the potential false negative rate in ExAC by assessing depth of sequencing coverage over PKD1 and PKD2. The mean coverage of PKD1 and PKD2 was 58.28× and 80.73×, respectively, and was variable for each exon (Supplementary Figure 2B and 2C, Supplementary Figure 3). As expected,3 exons 1 and 42 of PKD1, and exon 1 of PKD2, with high GC content >75%, had poor or no sequencing coverage (Supplementary Table 3). The power to detect variants is closely related to sequencing coverage, where >15× mean depth gives 97.5% sensitivity for detection of heterozygous single-nucleotide variants with exome sequencing.19 On average, 74% of PKD1 and 99% of PKD2 had >15× coverage across the cohort.
On balance, we are confident that the variants reported in ExAC for PKD1 are overwhelmingly real and thus represent a useful resource for interpreting variants in patients. We note that because poor quality variants have been filtered out of the ExAC database and given the variable coverage over some regions, there may be an ascertainment bias, making our results conservative.
PKD1 and PKD2 variants identified in ExAC
We assessed the frequency of genetic variants affecting PKD1 or PKD2 reported within the ExAC cohort of 60,706 unrelated individuals. We identified 4750 and 672 unique variants a total of 312,302 and 86,315 times in PKD1 and PKD2 respectively. The majority (70–75%) of these variants were either protein coding or in the essential splice region. Seventy-five percent of the reported unique PKD1 variants and 85% of unique PKD2 variants were nonsynonymous. There were 25 protein-truncating variants in PKD1 and PKD2 observed.
Protein-truncating PKD1 and PKD2 variants in ExAC
Loss-of-function variants in ADPKD are considered to be disease-causing.1 We thus examined the protein-truncating variants in more detail. In ExAC, there were 12 protein-truncating variants reported in PKD1 and 13 in PKD2 (Table 1). We manually reviewed the published evidence supporting all truncating variants in ExAC and also classified as pathogenic in the PKDB. All had been previously reported as pathogenic in small ADPKD pedigrees, in patients with clear phenotype, though segregation had not been possible in all cases.7,20,21 For example the PKD1 p.Arg4228Ter truncating variant is reported in eight different small pedigrees (<4 individuals) in PKDB and in one 65-year-old patient in ExAC, with the published reports describing patients with ESRF in their 30s–50s (refs. 22,23,24).
We investigated the possibility that individuals in ExAC with protein-truncating variants were somatic mosaics with reduced mutant load in the kidney and therefore not expected to have fully penetrant disease.25,26,27 We recorded the variant allele frequency for each protein-truncating variant in PKD1 and PKD2 from the ExAC browser’s interactive IGV.js browser.28,29 The minimum variant allele frequency was 33% and the maximum 52%, suggesting somatic mosaicism is unlikely to be an explanation for the presence of these variants in a control data set (Supplementary Table 4).
Curated PKD1 and PKD2 variants reported in PKDB
The PKDB contains expert-curated records of genetic variants observed in ADPKD patients, including single-nucleotide variants and small and large deletions and duplications. These variants are classified as “definitely pathogenic,” “highly likely pathogenic,” “likely pathogenic,” “likely hypomorphic,” “indeterminate,” and “likely neutral.” All protein-truncating variants and large deletions or duplications in PKD1 or PKD2 are classified as “definitely pathogenic.” Missense variants, in-frame deletions and duplications, and synonymous variants are classified as either “highly likely pathogenic,” “likely pathogenic,” “likely hypomorphic,” “indeterminate,” or “likely neutral” based on species conservation, in silico predictions, functional assays, and previous reports of pathogenicity.
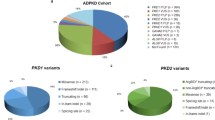
We first considered the number of unique variants found in common between PKDB and ExAC and then the total allele count of each variant. There were 2097 unique variants in PKD1 reported in the PKDB, and of these, 516 are also reported in ExAC, none of which overlapped with potential pseudogene false positive variants identified above. A total of 34 different variants were classified as definitely pathogenic (n = 5), highly likely pathogenic (n = 4), and likely pathogenic (n = 25) (Figs. 1a, b, 2a; Supplementary Table 5). The remaining 482 variants were classified as either likely hypomorphic (n = 5), indeterminate (n = 62), and likely neutral (n = 415). A number of these variants were identified in multiple participants in ExAC, which is represented by the allele count in Fig. 2c. In total, we observed 363 PKD1 records in ExAC, of variants that have been classified in the PKDB as definitely pathogenic, highly likely pathogenic, or likely pathogenic (Fig. 2c; Supplementary Table 5 and Supplementary Figure 4).

PKD1 and PKD2 variants in ExAC and PKDB. The number of unique variants identified in both ExAC and PKDB in PKD1 and PKD2. a Number of unique variants in PKD1 in each database and the number of different variants in common between databases. b Pathogenicity score (assigned by the PKDB) for each variant that is common between the two data sets in PKD1. c, d The same data for PKD2. ExAC Exome Aggregation Consortium, PKDB Polycystic Kidney Disease Database.

Location and allele count of disease-causing variants in common between PKDB and ExAC. a, b PKD1 and PKD2 disease-causing variants common between PKDB and ExAC, respectively, using ProteinPaint.40 The dotted line in (a) highlights exons 1–33 with pseudogene homology. c, d Allele count in the ExAC cohort of only the disease-causing PKD1 variant in common between the two databases; (d) depicts the allele count of all PKD2 variants common between the PKDB and ExAC. Variants identified in ExAC but previously classified in PKDB as likely neutral are not included in these figures. Here, we consider disease-causing as those previously classified in PKDB as definitely pathogenic, highly likely pathogenic, or likely pathogenic. ExAC Exome Aggregation Consortium, PKDB Polycystic Kidney Disease Database.
There were 264 unique PKD2 variants reported in PKDB, of which 47 were also reported in ExAC (Fig. 1c, d). Ten of these 47 variants were curated as definitely pathogenic (n = 5), highly likely pathogenic (n = 1), and likely pathogenic (n = 4), with one additional variant annotated as likely hypomorphic (Figs. 1c,d, 2b; Supplementary Table 4). A number of these variants were reported in more than one participant in ExAC, such that there were 42 PKD2 entries in ExAC of variants that have been classified as definitely pathogenic, highly likely pathogenic, or likely pathogenic in PKDB (Fig. 2d).
Expected frequency of disease-causing variants in ExAC
In a random population of 60,706 people sequenced by exome sequencing, it would be expected that 42 (0.69 per 1000) would have disease-causing variants identified in PKD1 and PKD2 (Fig. 3). In calculating this figure, we made the conservative assumption that ExAC had the same disease prevalence of ADPKD to the general population (1 per 1000). We also accounted for the diminished power to detect variants in all exons of PKD1 and PKD2 by exome sequencing (i.e., 74% with depth >15×). Furthermore, we accounted for only 90% of patients having a diagnosis due to variants in PKD1 or PKD2 and that 97% of diagnoses would be from single-nucleotide variants or indels, as opposed to large deletions (3% [ref. 7]). Finally, we assumed 75% of disease-causing variants in ExAC would be due to PKD1 variants and 25% due to PKD2 variants (in published ADPKD cohorts, PKD1 variants account for 80% of disease1).

Expected and observed disease-causing variants in ExAC. Number of disease-causing variants expected in ExAC based on known prevalence of autosomal dominant polycystic kidney disease (ADPKD; see text for details), compared with number of disease-causing variants observed in ExAC.
If we consider all variants in ADPKD classified as definitely pathogenic, highly likely pathogenic, or likely pathogenic, as well as novel truncating variants in ExAC, as likely to be disease-causing, then we identified a total of 418 records in ExAC, suggesting a disease prevalence of up to 6.9 per 1000 (Fig. 3). If we exclude all likely pathogenic variants, then we identified a total of 40 records in ExAC, and a prevalence of 0.66 per 1000. Based on this more conservative definition of variants contributing to disease, this number matches the expected prevalence under the assumptions made above. This suggests that a number of the variants currently classified as likely pathogenic in the PKDB may be benign, hypomorphic, or weakly penetrant, when population data is taken into account.
We calculated the maximum credible allele frequency for any individual disease-causing variant as 6.25 × 10−6, which is a maximum allele count of 5, in the ExAC population (see Methods). There were 46 different PKD1 and 8 different PKD2 variants that were each reported in at least 5 participants in ExAC (Supplementary Table 5). We suggest that these 54 variants are too frequently identified in the population database to be independently disease-causing and should be considered for reclassification.
To assess whether variant carriers in ExAC were young and thus potentially presymptomatic carriers of ADPKD, we analyzed the age distribution for all participants with any variant in PKD1 and PKD2 and compared this with the age distribution of ExAC participants with truncating variants in PKD1 and PKD2. The ages for the participants in ExAC with protein-truncating variants were similar to those with other types of variants (Fig. 4).

Ages of participants in ExAC with PKD1 and PKD2 disease-causing variants. Violin plot of the age of participants in the ExAC database with variants in PKD1 or PKD2. Also shown are the ages of participants in ExAC with variants that are classified as disease-causing in the PKDB and the ages of those ExAC participants with truncating variants. ExAC—variants in ExAC; ExAC and PKDB—variants identified in ExAC and also classified in the PKDB as definitely pathogenic, highly likely pathogenic, or likely pathogenic; truncating in ExAC—truncating variants in ExAC; red line—mean age of renal failure published for each group.1, 3, 9 ExAC Exome Aggregation Consortium, PKDB Polycystic Kidney Disease Database.
Discussion
This study demonstrates that population data sets comprised of exome sequencing data can be reliably applied in ADPKD to help refine variant classification, despite pseudogene homology. Initial examination of the ExAC population database shows that there are more disease-causing variants (defined in the PKDB as pathogenic, highly likely pathogenic, and likely pathogenic) than would be expected for the prevalence of ADPKD. Prior to the recent availability of large control data sets such as ExAC, interpretation of variants, particularly missense variants, was based largely on in silico analysis tools, segregation in small pedigrees, and previous reports. When population data are combined with these parameters, a number of variants currently classified as disease-causing in the PKDB may be reclassified as unlikely disease-causing based on their frequency in the population database. If only variants classified in the disease database as truncating or highly likely pathogenic are considered, the variant burden in ExAC matches the expected prevalence. Importantly, if a number of currently known PKD1 or PKD2 variants are reclassified as likely benign, this increases the proportion of patients with an ADPKD phenotype and no known disease-causing variant. Reanalysis in these cases may identify pathogenic variants in previously unexamined regions of PKD1 or PKD2 (such as promoter or intronic regions); newly described genes, such as GANAB; or result in gene discovery.30 This finding has important implications for individuals with disease, for whom genetic information is used to guide family planning, access to treatments, and prognosis.
As access to large data sets of genomic information has increased, the reassessment of previously reported pathogenic variants, which were reported using the best available evidence at the time, has been demonstrated in other disease groups.31 These findings support recent guidelines relating to cautious use of in silico pathogenicity prediction tools in the assessment of disease-causing variation.8 Also highlighted is the difficulty of adequate wide segregation in an adult-onset autosomal dominant disease, in which often only probands or small pedigrees are available for analysis.8
An additional interesting finding is the number of patients, of broadly distributed age, with protein-truncating or highly likely pathogenic variants in ExAC. If we make the conservative assumption that ExAC is a cohort of individuals with typical population risk for ADPKD, then the number of identified and expected individuals is approximately equal, as has been reported previously for variants in PKD2 (ref. 13). If however, the ExAC cohort is biased toward healthy individuals, as has been reported by the ExAC curators9 and the previous PKD2 investigation,13 then there is an excess of apparently healthy individuals carrying loss-of-function variants, which would otherwise be reported as pathogenic by most clinical laboratories. This suggests that some truncating variants in ADPKD may have reduced penetrance, and that some should be considered disease-predisposing rather than disease-causing. To validate this will require additional follow-up with extensive phenotype data.
Interestingly, the truncating variants identified were enriched toward the 3’ end of PKD1, perhaps suggesting that truncating variants toward the end of the protein may be variably penetrant, though this requires further extensive study. Notably, truncating variants identified in the last exons of PKD1 and PKD2 have been reported as definitely pathogenic in the PKDB and in recent literature.7,32 The most 3’ truncating variant reported in PKDB as pathogenic is at codon 4276/4304 of PKD1, and at codon 949/969 of PKD2 (ref. 12) (Fig. 2a, b).
The power of variant databases to improve variant classification has been demonstrated in other disease groups. A 2016 study by Minikel et al. utilized the ExAC data set to interpret variants in the adult-onset disorder prion disease.10 The group found that there were more than 30 times more variants in the population data sets than was expected by known prevalence of the disease. They concluded that there was significant variability in lifetime risk of developing disease based on the particular variant inherited and its prevalence in control populations.10 Variants detected in multiple affected families and absent or at very low prevalence in the population data sets had 100% lifetime risk of developing disease, as compared with variants that had higher prevalence in population cohorts.10 Similar studies utilizing the ExAC data set to assess penetrance and variant pathogenicity have been performed in genetic adult-onset cardiomyopathy, genetic ventricular tachycardias, and schizophrenia and intellectual disability.33,34,35
A limitation of this study is that detailed phenotype information is not available for ExAC participants. We reviewed the inclusion and exclusion criteria for each study that comprises the ExAC cohort and found no suggestion that the database is enriched for kidney disease.9 Previous studies have also demonstrated that the database is not enriched for pathogenic variants.36 Based on our current understanding of ADPKD, patients with PKD1 truncating variants have median age of onset of ESRF in their mid 50s.1 We thus used the age of each ExAC individual as a surrogate phenotype. We demonstrated that the ages of participants in ExAC with PKD1 or PKD2 protein-truncating variants were evenly distributed across age brackets, suggesting that the findings are not skewed to participants of a young age who are yet to manifest a phenotype (Fig. 4). Another potential limitation is that whilst the majority of the individuals in the ExAC cohort9 and PKDB registry are of European ancestry, any substantial differences in ancestry may lead to subtle biases, and potentially more rare alleles in cases relative to controls.
There is ascertainment bias in ADPKD literature, given probands are identified when they manifest clinically significant disease. It is possible that ADPKD is more prevalent than currently understood and that patients with subclinical disease are currently not identified. This is supported by autopsy studies that report ADPKD at a prevalence more frequent than 1/1000 (refs. 37,38,39). Investigating the true population prevalence of ADPKD will require further study of large cohorts of unselected individuals, with the ability to undertake follow-up clinical assessment in individuals with truncating or previously reported disease-causing variants.
A major challenge in genetic sequencing in ADPKD is the presence of six pseudogenes that share approximately 97% sequence homology with two-thirds of the PKD1 gene.5 Our analysis of the exome sequencing data from the ExAC data set demonstrates that depth of coverage over PKD1 is reduced in the pseudogene-homologous region. However, analysis of variants reported in this region shows good mapping quality. Therefore, although the exome sequencing method used by ExAC does not adequately cover PKD1 for the purposes of diagnostic sequencing, our analysis demonstrates that the variants reported in the database are likely to be real. This adds value to the interpretation of variants reported in the ExAC database; however, the absence of a variant in the ExAC database, in the PKD1 pseudogene-homologous region, is a less meaningful finding given the variable coverage in this region. This is relevant as the absence of a variant in control data sets is a pathogenicity-criteria in the current American College of Medical Genetics and Genomics (ACMG) guidelines.8 These are valuable findings for those interpreting variants in ADPKD, both in the diagnostic and research setting, as the majority of control data sets currently utilize exome sequencing data.
Our study emphasizes the complexity of variant interpretation in ADPKD and the challenge of estimating the likelihood of a particular sequence variant resulting in clinically significant disease in an individual. Our findings demonstrate how classification of variants has evolved with rapidly increasing amounts of data and highlight the value of reviewing previous variant findings. These findings are not unique to ADPKD and likely applicable to other, particularly adult-onset, autosomal dominant genetic diseases. The findings of our study reinforce the value of international collaboration to ensure that disease and population databases are inclusive and well curated. These challenges will be ongoing as our genetic knowledge continues to increase and raises wider issues regarding resourcing to allow for review of previously classified variants, for both diagnostic and research laboratories. We demonstrate that ExAC data can be used in regions of pseudogene homology, because the presence of a variant is likely to be true. However, given reduced coverage, our data indicate that the absence of a variant in a homologous region is less powerful. Our study demonstrates that some previously reported ADPKD variants are unlikely to be disease-causing and highlights the complexity of predicting disease severity in ADPKD, even with genetic information—this is important knowledge for clinicians counseling patients and for researchers striving to better understand the pathogenesis of this common monogenic condition.
References
Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–1013.
Cornec-Le Gall E, Audrezet MP, Rousseau A, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27:942–951.
Mallawaarachchi AC, Hort Y, Cowley MJ, et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur J Hum Genet. 2016;24:1584–1590.
Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol. 2018;29:13–23.
Bogdanova N, Markoff A, Gerke V, et al. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics. 2001;74:333–341.
Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160.
Audrézet M-P, Cornec-Le Gall E, Chen J-M, et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239–1250.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Minikel E, Vallabh S, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9.
Natarajan P, Gold N, Bick A, et al. Aggregate penetrance of genomic variants for actionable disorders in European and African Americans. Sci Transl Med. 2016;8:364ra151.
The Mayo Clinic, ADPKD Mutation Database: PKDB. http://pkd.mayo.edu. Accessed 20 May 2016.
Cornec-Le Gall E, Audrézet M-P, Renaudineau E, et al. PKD2-Related autosomal dominant polycystic kidney disease: prevalence, clinical presentation, mutation spectrum, and prognosis. Am J Kidney Dis. 2017;70:476–485.
McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760.
Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079.
Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158.
Meynert A, Ansari M, FitzPatrick D, Taylor M. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics. 2014;15:247–258.
Carrera P, Calzavara S, Magistroni R, et al. Deciphering variability of PKD1 and PKD2 in an Italian cohort of 643 patients with autosomal dominant polycystic kidney disease (ADPKD). Sci Rep. 2016;6:srep30850.
Trujillano D, Bullich G, Ossowski S, et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol Genet Genomic Med. 2014;2:412–421.
Hoefele J, Mayer K, Scholz M, Klein HG. Novel PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2011;26:2181–2188.
Peral B, Gamble V, Strong C, et al. Identification of mutations in the duplicated region of the polycystic kidney disease 1 gene (PKD1) by a novel approach. Am J Hum Genet. 1997;60:1399–1410.
Peral B, San Millan J, Ong A, et al. Screening the 3’ region of the polycystic kidney disease 1 (PKD1) gene reveals six novel mutations. Am J Hum Genet. 1996;58:86–96.
Tarailo-Graovac M, Zhu JYA, Matthews A, van Karnebeek CDM, Wasserman WW. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet Med. 2017;19:1300–1308.
Pagnamenta AT, Lise S, Harrison V, et al. Exome sequencing can detect pathogenic mosaic mutations present at low allele frequencies. J Hum Genet. 2011;57:70–72.
Simons C, Dyment D, Bent SJ, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 2017;140:3105–3111.
Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26.
Karczewski KJ, Weisburd B, Thomas B, et al. The ExAC browser: displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017;45:D840–D845.
Porath B, Gainullin VG, Gall EC-L, et al. Mutations in GANAB, encoding the glucosidase IIa subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016;98:1193–1207.
Bell C, Dinwiddie DL, Miller N, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:1–16.
Audrezet MP, Corbiere C, Lebbah S, et al. Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27:722–729.
Akinrinade O, Koskenvuo JW, Alastalo T-P. Prevalence of titin truncating variants in general population. PLoS ONE. 2015;10:e0145284.
Ropers HH, Wienker T. Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. Eur J Med Genet. 2015;58:715–718.
Paludan-Müller C, Ahlberg G, Ghouse J, et al. Integration of 60,000 exomes and ACMG guidelines question the role of catecholaminergic polymorphic ventricular tachycardia-associated variants. Clin Genet. 2017;91:63–72.
Song W, Gardner SA, Hovhannisyan H, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: insights of relevance to variant classification. Genet Med. 2016;18:850–854.
Barakat A, Drougas J. Occurrence of congenital abnormalities of kidney and urinary tract in 13,775 autopsies. Urology. 1991;38:347–350.
Iglesias CG, Torres VE, Offord KP, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-80. Am J Kidney Dis. 1983;2:630–639.
Chan KW. Adult polycystic kidney disease in Hong Kong Chinese: an autopsy study. Pathology. 1993;25:229–232.
Zhou X, Edmonson MN, Wilkinson MR, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016;48:4–6.
Acknowledgements
M.J.C. is supported by a New South Wales (NSW) Health Early-Mid Career Fellowship, Cancer Institute NSW Early Career Fellowship (13/ECF1-46), and the Kinghorn Foundation. A.C.M. is supported by the Royal Australasian College of Physicians (RACP) Jacquot Foundation. We are grateful to Daniel MacArthur and Anne O’Donnell from the Broad Institute for providing the age data for the ExAC participants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Mallawaarachchi, A.C., Furlong, T.J., Shine, J. et al. Population data improves variant interpretation in autosomal dominant polycystic kidney disease. Genet Med 21, 1425–1434 (2019). https://doi.org/10.1038/s41436-018-0324-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0324-x
Keywords
This article is cited by
-
Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank
Nature Genetics (2022)
-
Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing
European Journal of Human Genetics (2021)
