
Abstract
Purpose
Neurofibromatosis type 1 (NF1) is characterized by a highly variable clinical presentation, but almost all NF1-affected adults present with cutaneous and/or subcutaneous neurofibromas. Exceptions are individuals heterozygous for the NF1 in-frame deletion, c.2970_2972del (p.Met992del), associated with a mild phenotype without any externally visible tumors.
Methods
A total of 135 individuals from 103 unrelated families, all carrying the constitutional NF1 p.Met992del pathogenic variant and clinically assessed using the same standardized phenotypic checklist form, were included in this study.
Results
None of the individuals had externally visible plexiform or histopathologically confirmed cutaneous or subcutaneous neurofibromas. We did not identify any complications, such as symptomatic optic pathway gliomas (OPGs) or symptomatic spinal neurofibromas; however, 4.8% of individuals had nonoptic brain tumors, mostly low-grade and asymptomatic, and 38.8% had cognitive impairment/learning disabilities. In an individual with the NF1 constitutional c.2970_2972del and three astrocytomas, we provided proof that all were NF1-associated tumors given loss of heterozygosity at three intragenic NF1 microsatellite markers and c.2970_2972del.
Conclusion
We demonstrate that individuals with the NF1 p.Met992del pathogenic variant have a mild NF1 phenotype lacking clinically suspected plexiform, cutaneous, or subcutaneous neurofibromas. However, learning difficulties are clearly part of the phenotypic presentation in these individuals and will require specialized care.
Similar content being viewed by others


Introduction
Neurofibromatosis type 1 (NF1, MIM 162200) is an autosomal dominant disorder affecting 1 in every 2000 to 3000 births with age-dependent penetrance and highly variable inter- and intrafamilial expressivity.1 The main clinical signs of NF1 include café-au-lait macules (CALMs), skinfold freckling, Lisch nodules, neurofibromas, optic pathway gliomas (OPGs), and/or specific skeletal abnormalities, such as sphenoid wing or tibial dysplasia. According to the diagnostic criteria established by the National Institutes of Health (NIH), the presence of at least two of the aforementioned features (or a single clinical symptom with a first-degree relative meeting NIH criteria) is sufficient for a clinical diagnosis of NF12. The NIH diagnostic criteria are highly specific and sensitive in NF1-affected adults as nearly all have cutaneous and/or subcutaneous neurofibromas.3 Examples of individuals consistently presenting only with typical pigmentary manifestations (multiple CALMs with/without freckling) without externally visible plexiform, cutaneous, or subcutaneous neurofibromas are these heterozygous for the NF1 deletion p.Met992del and missense pathogenic variants at residue p.Arg18094,5,6. Because NF1 is a progressive disorder with phenotypic manifestations increasing with age, the development of serious complications still needs consideration in individuals with p.Met992del until more data, especially in adults, have become available. The association of p.Met992del with a mild phenotype was originally established based on the analysis of 21 unrelated probands and 26 affected relatives, with only 19 individuals ≥19 years old.4 Larger data sets, preferably of adults, are required to confirm the mild clinical course in these individuals and identify rare complications.
We describe in this study detailed phenotypic characterization of an additional 135 individuals from 103 unrelated families carrying the NF1 p.Met992del pathogenic variant. Besides confirming the absence of superficial neurofibromas, we demonstrate that some complications, such as nonoptic brain tumors and cognitive impairment and/or learning disabilities, are present in a significant number of these individuals (4.8% and 38.8%, respectively). We also provide proof of the existence of low-grade pilocytic astrocytomas with the NF1 p.Met992del pathogenic variant and loss of heterozygosity (LOH) at p.Met992del and three intragenic NF1 microsatellite markers in three lesions from a single NF1-affected individual, establishing these as NF1-associated tumors.
Materials and methods
A total of 135 individuals from 103 unrelated families were referred to the Medical Genomics Laboratory at University of Alabama at Birmingham (UAB; 74 probands and 27 affected relatives) and collaborating centers (EUR; 29 probands and 5 affected relatives) for NF1 molecular testing (see details in Table S1). All were confirmed as having the same NF1 3-bp in-frame deletion c.2970_2972del, leading to loss of a methionine at codon 992 (p.Met992del), except for a 14-month-old girl heterozygous for the NF1 c.2974_2976del, also resulting in p.Met992del (UAB-R7536). Comprehensive NF1 molecular analysis with an RNA-based approach was performed in the Medical Genomics Laboratory as previously described,6,7 with LRG_214 and NM_000267.3 used as the reference sequences.
Clinical data were collected as previously reported6,7 at the time of genetic testing with data reverified by referring physicians for accuracy and/or updating, when available. As magnetic resonance imaging (MRI) is not routinely performed in individuals without clinical signs/suspicion for spinal neurofibromas and/or OPGs, presence of internal neurofibromas and asymptomatic OPGs could not be excluded in all cases. If ≥2 Noonan-like features (i.e., short stature, facial dysmorphism [low-set ears, hypertelorism, downslanted palpebral fissures, midface hypoplasia, ptosis, and/or webbed neck] or cardiac abnormality [pulmonic stenosis]) were present, an individual was classified as having Noonan-like phenotype. Short stature and macrocephaly were defined as previously described.6,7
We compared the phenotypes of individuals with the NF1 p.Met992del pathogenic variant with the cohorts of individuals heterozygous for the NF1 missense pathogenic variants affecting codons 1809 and 844–848 5,6,7,8,9,10, and previously described large-scale cohorts with “classic” NF111,12,13,14,15,16,17,18,19,20,21,22,23,24. Two-tailed Fisher’s exact test with P values adjusted for multiple comparisons using Benjamini–Hochberg (B–H) procedure with false discovery rates (FDR) at 0.05 and 0.01 was applied (GraphPad software; Table S2).
This study was approved by the institutional review boards of all participating institutions offering clinical genetic testing.
Results
Clinical description of the studied group
Among 103 unrelated probands, 38/103 (36.9%) were familial and 43/103 (41.8%) were sporadic cases, with 6/43 individuals proven to have a de novo variant, but no formal paternity/maternity testing was performed. Two individuals with proven de novo p.Met992del (UAB-R6151 and UAB-R2586) had one parent reported to have 1–5 CALMs with/without freckling; thus gonosomal mosaicism or the presence of a different independent NF1 pathogenic variant cannot be excluded in these families (Table S1). For 22/103 (21.4%) cases family history was unknown, including three adopted probands.
Ninety-seven of 131 individuals (74.1%) fulfilled the NIH diagnostic criteria, but only 76/131 (58%) if family history was excluded as a criterion (Table 1). Of the 55 cases not fulfilling the NIH diagnostic criteria after excluding the family history, 31 had complete clinical information including the ophthalmological results for the presence/absence of Lisch nodules and symptomatic OPGs (Table S1 and Table S3), including 11/31 (35.5%) children ≤8 years old, 10/31 (32.3%) individuals between 9 and 18 years old, and 10/31 (32.3%) adults over 19 years old. Five of 31 individuals had <6 CALMs as the only clinical feature (Table S3).
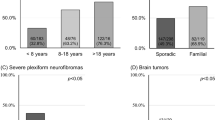
The presence of >5 CALMs and freckling was observed in 119/135 (88.2%) and 73/124 (58.9%) cases, respectively. Fifty-two of 85 individuals ≥9 years old had both pigmentary manifestations, while Lisch nodules were observed in 12.9% (13/101) of the studied cohort, including 14.9% (10/67) ≥9 years old. No symptomatic OPGs were found herein (0/123); however, of 41 asymptomatic individuals receiving brain MRI screening, a single bilateral OPG located in chiasm and optic nerves was reported (EUR-R1).
No histopathologically confirmed externally visible plexiform (0/128, including 0/84 individuals ≥9 years old), cutaneous (0/128, including 0/38 individuals ≥19 years), or subcutaneous neurofibromas (0/124, including 0/36 individuals ≥19 years) were found. In 14 cases ≥14 years a single or a few cutaneous or subcutaneous lesions suspected to be neurofibromas were observed (Table S1 and Table S4). Importantly, for five individuals the diagnosis was “lipoma” instead after detailed clinical and/or histopathological analysis. Symptomatic spinal neurofibromas were absent in all 118 individuals and the presence of asymptomatic spinal tumors was excluded by MRI in 13 cases (with 12/13 individuals ≥9 years old).
Twenty-one of 125 individuals (16.8%) had skeletal abnormalities (Table 1 and Table S1). Besides scoliosis (11/125 all ages, but 5/38 ≥19 years) and pectus anomalies (9/125), other skeletal abnormalities included rib abnormality, bilateral club feet, dysplastic vertebrae, mild winging of the scapula and kyphosis (each observed in a single individual).
Nonmalignant tumors, not including OPGs and neurofibromas, were identified in 12/126 (9.5%) individuals and included astrocytomas (n = 3, one tumor was described as having features of astrocytoma and dysembryoplastic neuroepithelial tumor [DNET], see details in Table S1), oligodendroglioma (n = 1), lipomas (n = 5), angiolipoma (n = 1), hypothalamic glioma (n = 1), brain tumor with hamartomatous aspect by MRI of the encephalon (n = 1), and craniopharyngioma (n = 1) (Table 1). In addition, one individual had a history of a neuroblastoma, but detailed follow-up was not available (Table S1 and Table S5). No breast cancer was observed in the studied cohort (0/20 women ≥19 years). Of particular interest was a 42-year-old male with no clinical signs of NF1 (UAB-R5571-F) and no history of prior irradiation, but with three surgically removed juvenile pilocytic astrocytomas, one located in the brainstem and two in the top left side of the brain (Table S1). He was molecularly diagnosed as being heterozygous for NF1 p.Met992del after diagnosis was made in his son who had >5 CALMs, left inguinal freckling, macrocephaly, and abnormal development (Fig. S1). Morphologically, an astrocytic neoplasm composed of two growth patterns was observed, i.e., areas with a fibrillary arrangement and Rosenthal fibers and areas with a pattern similar to that seen in oligodendroglioma; there was no significant pleomorphism, increased mitotic activity, or necrosis. Molecular analysis revealed the NF1 pathogenic variant c.2970_2972del with LOH at c.2970_2972del and all three intragenic NF1 microsatellite markers tested in all three astrocytomas, confirming these as NF1-associated tumors.
Fifty of 129 case subjects (38.8%) had an abnormal development with at least one of the following forms of cognitive impairment or learning disabilities: learning difficulties (n = 38), developmental delay (n = 10), speech delay (n = 15), motor delay (n = 7), autism spectrum (n = 1), or psychiatric problems (n = 2). Of these, three individuals (UAB-R1873, UAB-R1873-M, and UAB-R6975) carried known additional genomic imbalances: 1q21.1 deletion (MIM 612474), 12q23.2 duplication, and 15q13.3 duplication (Table S1 and Fig. S2). In addition, array comparative genomic hybridization (aCGH) analysis revealed the presence of a 12.6-kb-sized deletion on 16p13 and 226.9 kb-sized duplication on 14q32.13 of unclear clinical significance in a single individual (UAB-R8603) (Table S1). Noonan-like features were found in 15/119 (12.6%) individuals, including the presence of pulmonic stenosis in four children ≤8 years old. In all 10 individuals with Noonan-like phenotype referred to UAB no pathogenic or likely pathogenic variants in 16 Noonan-related disorder genes (see details in Table S1) were identified. Other cardiovascular abnormalities included hypertension, double aortic arch with vascular ring, ventricular tachycardia, sinus arrhythmia and atypia of repolarization, and atrial septal defect (each observed in a single individual). Short stature and macrocephaly were found in 11/71 (15.5%) and 26/87 (29.9%) of case subjects, respectively.
Comparison of clinical features in individuals heterozygous for NF1 p.Met992del with cohorts of individuals with NF1 missense pathogenic variants affecting codons 1809 and 844–848 and with “classic” NF1 phenotype
Individuals with p.Met992del had Lisch nodules significantly less often and no externally visible plexiform neurofibromas, cutaneous and subcutaneous neurofibromas, compared with the cohort of individuals with the NF1 missense pathogenic variants at residues 844–848 and previously reported “classic” NF1 population (all P < 0.0001, statistically significant after B–H correction at FDR 0.01; Table 2). Importantly, no individuals described in this and previous studies had visible plexiform neurofibromas (0/127 ≥9 years) or histopathologically confirmed cutaneous (0/59 ≥19 years) or subcutaneous neurofibromas (0/37 ≥19 years) (Table S6).4,25,26 Similar to the NF1 p.Arg1809 cohort, no symptomatic OPGs were observed in the studied group. In the current study, none of the individuals showed symptomatic spinal neurofibromas (Table 1), although in the original report a single symptomatic spinal tumor was found;4 nevertheless, even combined that remains significantly less prevalent than in the cohort of individuals with the NF1 missense pathogenic variants at residues 844–848 (P < 0.0001, statistically significant after B–H correction at FDR 0.01).
The mild phenotype without any externally visible neurofibromas observed in the current study is therefore similar to that of individuals with the NF1 p.Arg1809 missense pathogenic variants, except for the presence of brain tumors being more prevalent in the p.Met992del-positive cohort (4.8% versus 0.8%), but this difference was not statistically significant (P = 0.0640).
The prevalence of skeletal abnormalities was similar as in the NF1 p.Arg1809 cohort and “classic” NF1 population (17.4% versus 16.7% versus 15.2%, respectively), but lower than in the cohort of individuals with the NF1 missense pathogenic variants at residues 844–848 (P = 0.0016, statistically significant after B–H correction at FDR 0.05).
Noonan-like features and pulmonic stenosis were much more prevalent in the studied group compared with the general NF1 population (P < 0.0001 and P = 0.0009, respectively, both statistically significant after B–H correction at FDR 0.01). However, compared with the p.Arg1809 cohort, individuals with p.Met992del had Noonan-like phenotype and short stature statistically less often (P < 0.0001 and P = 0.0056, statistically significant after B–H correction at FDR 0.01 and 0.05, respectively; Table 2). In addition, macrocephaly was observed less frequently than in the cohorts of individuals with the NF1 missense pathogenic variants at residues 844–848 and “classic” NF1 clinical presentation (P = 0.0267 and P = 0.0111, respectively, not statistically significant after B–H correction at FDR 0.05; Table 2). Finally, individuals with p.Met992del had a significantly lower prevalence of cognitive impairment and/or learning disabilities compared with the cohorts of individuals with the NF1 missense pathogenic variants at codon 1809 and the general NF1 population (P = 0.0018 and P = 0.0082, both statistically significant after B–H correction at FDR 0.05; Table 2). Comparing the current cohort with the initially reported p.Met992del individuals,4 the overall frequency of abnormal development with/without learning disabilities was significantly higher in the current study (38.8% versus 17%; P = 0.0066; Table S7), even after excluding the individuals with genomic imbalances that may have a modifying effect (36.8% versus 17%; P = 0.0160; Table S7).
Discussion
A renewed interest in NF1 genotype–phenotype correlations is emerging, especially with the rapid development and accessibility of genomic technology, given the recent identification of four clinically significant genotype–phenotype correlations.4,5,6,7,27 The constitutional NF1 microdeletion and NF1 missense pathogenic variants affecting codons 844–848 are important risk factors for severe presentation, including a high prevalence of plexiform neurofibromas at an earlier age, dysmorphic facial features and global developmental delay with/without learning disabilities and an increased lifetime risk for malignant peripheral nerve sheath tumors (MPNSTs) in individuals with the NF1 microdeletion,27 and a significant increase in number of plexiform and symptomatic spinal neurofibromas, symptomatic OPGs, skeletal abnormalities, and malignant neoplasms in individuals with the NF1 missense pathogenic variants at residues 844–8487. Furthermore, a mild clinical presentation lacking any externally visible plexiform, cutaneous, or subcutaneous neurofibromas is observed in NF1-affected individuals heterozygous for the NF1 missense pathogenic variants at codon 18095,6 or the 3-bp in-frame NF1 deletion, c.2970_2972del (p.Met992del).4 Because the latter genotype–phenotype correlation was established on a limited number of NF1-affected adults (19/47), an update of this initial intragenic genotype–phenotype correlation is important for clinical practice.
The frequency of the p.Met992del pathogenic variant in the NF1 mutation–positive unrelated individuals from the UAB cohort is ~0.9% (74/8400), making it one of the most common recurring variants observed in the UAB database. This variant was reported in publicly available disease databases (last accessed July 2018), such as the Leiden Open Variation Database (LOVD; 28 times, 27/28 and 1/28 classified as pathogenic and likely pathogenic, respectively), ClinVar (7 times, all classified as pathogenic) and the Human Gene Mutation Database (HGMD; classified as pathogenic) and absent in population databases, 1000 Genomes, and the Exome Variant Server (EVS), except for a single report in the Genome Aggregation Database (gnomAD; the variant’s frequency in all populations is 0.00041%), and completely segregated with the disorder in 14 affected individuals from 12 unrelated families and was proven to be de novo in six probands. As such, the NF1 deletion, c.2970_2972del (p.Met992del), should undoubtedly be classified as pathogenic according to current recommendations.28
Cutaneous and subcutaneous neurofibromas are benign tumors located on or just under the skin, typically developing around puberty, and almost all NF1-affected adults have at least several of them.16 Over the course of time, the number of neurofibromas usually increases, varying from hundreds to thousands. Plexiform neurofibromas may be recognized earlier as they may occur congenitally, but most of them grow slowly, with externally visible plexiform neurofibromas becoming apparent in the early years of life. Plexiform neurofibromas arise in peripheral nerves and are clinically suspected in 15–30% of the NF1-affected general population, but the prevalence of the internal tumors is higher.3,11,18,29,30,31 Moreover, plexiform and subcutaneous neurofibromas are associated with an increased lifetime risk for the development of MPNSTs, resulting in significant morbidity for these individuals.32,33,34 From a clinical point of view, it is important to identify early those individuals with an increased risk to develop malignancies so as to provide them personalized management and genetic counseling.
In the current study, we confirmed the paucity of superficial plexiform (0/84 ≥9 years) and cutaneous neurofibromas (0/38 ≥19 years), in line with the original report.4 None of the individuals had externally visible plexiform neurofibromas or histopathologically confirmed cutaneous or subcutaneous neurofibromas (all P < 0.0001, statistically significant at FDR of 0.01 after B–H correction when compared with the cohort of individuals with the NF1 missense pathogenic variants affecting codons 844–848 and the “classic” NF1-affected population, Table 2). Combining data from this and previous studies4,25,26 for plexiform (0/127 ≥9 years), cutaneous, and subcutaneous neurofibromas (0/59 and 0/37 ≥19 years, respectively), we estimate in post hoc power calculation that these sample sizes would allow to detect the presence of plexiform, cutaneous, and subcutaneous neurofibromas with a prevalence of at least 3%, 7%, and 10%, respectively with a power of 95%. However, we cannot speculate about the risk for internal neurofibromas in this cohort as MRI screening was not routinely done in most of the asymptomatic individuals.
The presence of possible few cutaneous or subcutaneous lesions was initially mentioned on the phenotypic checklist in 14 individuals ≥14 years, but upon further detailed clinical evaluation by NF1 experts and/or histopathological analysis the lesions were diagnosed as lipomas in 5/14 individuals (for the remaining cases no follow-up was available) (see Comments in Table S1 and Table S4). Moreover, in none of the individuals were additional lesions observed over time, further supporting that these lesions were unlikely to be neurofibromas, because cutaneous and subcutaneous neurofibromas usually increase in number over the years. The overall prevalence of lipomas in the p.Met922del-positive individuals from this and a previous study4 was 5.5% (7/128); lipomas have also been observed in ~20% of individuals (21/115) with Legius syndrome (MIM 611431), another mild phenotype consisting of pigmentary spots only, caused by pathogenic variants in SPRED1 (MIM 609291).35
Besides neurofibromas and OPGs, NF1-affected individuals may develop other benign and malignant tumors, such as MPNSTs, rhabdomyosarcomas, leukemias, neuroblastomas, pheochromocytomas, gastrointestinal stromal tumors (GISTs), glomus tumors, and breast and/or ovarian cancer36 that depending on the clinical and histological grade may significantly increase mortality in the NF1 population.1 Nonoptic gliomas are one of the most common brain tumors in NF1-affected individuals, with an overall frequency greater than 100 times that in the general population.37 In the current study, we reported 6/126 (4.8%) individuals with nonoptic brain tumors, mostly low-grade and asymptomatic (Table S1 and Table S5). The prevalence in the studied cohort was similar to that recorded in the recent report on nonoptic gliomas by Sellmer et al.38 (24/562, 4.3%). Additionally, we confirmed the presence of the NF1 p.Met992del pathogenic variant with LOH in three astrocytomas from a single case (UAB-R5571-F), confirming these as NF1-associated tumors. Burgoyne et al.26 also demonstrated the occurrence of a germline p.Met992del along with a somatic NF1 p.Ser1407fs*21 pathogenic variant in an individual with multiple CALMs and mild axillary freckling who developed a low-grade GIST. Notwithstanding the above, brain tumors seem to be the most common complications in individuals heterozygous for the constitutional NF1 p.Met992del. Even though brain tumors in NF1 usually are low-grade lesions and have a more benign course than in the individuals without NF139, it is important that clinicians involved in the care of individuals with NF1 are aware of this complication.
The highly variable inter- and intrafamilial expressivity and age-dependency of most symptoms undoubtedly hampers the accurate NF1 clinical diagnosis, especially in infants and individuals with mosaic NF1. Indeed, nearly half of sporadic NF1-affected children do not fulfill the NIH diagnostic criteria by 1 year of age.3 In this study, we observed that 14/60 (23.3%) of individuals ≥9 years did not fulfill the NIH diagnostic criteria. However, these numbers increase dramatically if the family history is not taken into account (23/60, 38.3%), which necessarily applies to all sporadic cases (Table S8). An important reason why the p.Met992del-positive individuals do not fulfill NIH criteria is the absence of any externally visible neurofibromas, even in adults. As only 10/67 (14.9%) of individuals ≥9 years developed Lisch nodules (Table 1), a systematic ophthalmological examination for Lisch nodules may not be of major help in making an early clinical diagnosis in the p.Met992del-positive individuals. The cumulative advances in our understanding of NF1 prompt the need to consider if additional clinical signs, such as juvenile xanthogranulomas (JXG), nevus anemicus (NA), or choroidal abnormalities, as well as proof of a pathogenic variant, may help to establish an early NF1 diagnosis, especially in children.40 However, screening for JXG, NA, and choroidal abnormalities was not routinely done in the current study.
Cognitive impairment and/or learning disabilities are part of the phenotype associated with the NF1 p.Met992del, as these were present in 50/129 individuals (38.8%), including five children with severe global developmental delay and/or gross motor delay (UAB-R1542, UAB-R1873, UAB-R4613, UAB-R6975, and UAB-R4846). In some cases, aCGH analysis was performed in addition to the NF1 molecular analysis, identifying genomic imbalances that may have modifying roles (Table S1). One of these cases (UAB-R6975), a 9-year-old girl with serious neurocognitive issues (severely deficient verbal intellectual function, impaired expressive and receptive language delays, and features of autism spectrum disorder, generalized anxiety disorder, and dyspraxia), was found to have a constitutional 15q13.3 duplication, encompassing the CHRNA7 (MIM 118511) and OTUD7A (MIM 612024) genes (Table S1). CHRNA7 duplication is however of unknown significance, although cognitive impairment and psychiatric disorders have been observed in several families with such microduplications.41 On the other hand, another individual (UAB-R4613) also with very severe developmental delay (inability to read and write at age 17 and only processing basic information, such as following directions and completing small assignments) had a normal aCGH result. Nevertheless, compared with the initial study,4 abnormal development with/without learning disabilities was much more common in the current study (P = 0.0066; Table S7) and requires special attention.
Furthermore, Noonan-like phenotype and pulmonic stenosis were more frequent in the studied cohort compared with the general NF1 population (P < 0.0001 and P = 0.0009, respectively; both statistically significant at FDR of 0.01 after B–H correction); however, 2/15 individuals showed presence of the 1q21.1 microdeletion syndrome (Fig. S2), which is associated with some Noonan-like features. Pathogenic or likely pathogenic variants in PTPN11 (MIM 176876) and/or other Noonan-related disorder genes were ruled out in 13/15 cases with Noonan-like phenotype; two individuals (EUR-R5 and EUR-R19) were not tested (Table S1).
Although the initial genotype–phenotype report, demonstrating very mild phenotype in the NF1 p.Met992del-positive individuals, was published a decade ago,4 the biological reasons why these specific individuals do not develop any externally visible neurofibromas are still unclear. Methionine at codon 992 is surrounded by evolutionarily highly conserved amino acids, but no significant progress has been made toward better understanding of this region of the protein. Besides the well-understood role of the GAP-related domain, only a few functional studies have verified how other domains regulate the function of neurofibromin.42 Therefore, there is a need to increase efforts to develop functional assays to improve our understanding of the biological effect of this and other pathogenic variants. Understanding the molecular mechanisms whereby p.Met992del is not associated with the development of neurofibromas in NF1-affected individuals may help to identify new therapeutic targets.
The mild phenotype described herein with mainly pigmentary manifestations only is not limited to NF1 because several other conditions with overlapping features, especially Legius syndrome and Noonan syndrome with multiple lentigines (formerly called LEOPARD syndrome, MIM 151100), may be phenotypically/clinically indistinguishable from the p.Met992del or p.Arg1809 phenotypes. As such, the 2016 American Association for Cancer Research (AACR) Childhood Cancer Predisposition workshop recommends that children fulfilling one or more NIH diagnostic criteria have the NF1 diagnosis molecularly confirmed.43 Establishing a correct NF1 diagnosis, especially in young individuals who do not meet the clinical criteria, is crucial in determining appropriate clinical management. Though genotype–phenotype correlations are exceptions in NF1, the identification of clinically relevant genotype–phenotype correlations facilitates counseling and surveillance of a significant number of NF1 patients.
In conclusion, we clearly confirmed that a 3-bp in-frame deletion of the NF1 gene, c.2970_2972del (p.Met992del), is associated with a mild phenotype lacking externally visible plexiform, cutaneous, or subcutaneous neurofibromas. Through the analysis on a well-described cohort of 135 individuals (including 90 individuals ≥9 years) we bring to attention the significant risk for cognitive impairment, learning disabilities, and nonoptic brain tumors associated with this particular NF1 genotype. Therefore, clinicians specializing in the care of NF1-affected individuals should be aware of these complications, mostly occurring in oligosymptomatic individuals who may, nevertheless, require personalized attention for pathogenic variant-specific complications.
URLs
1000 Genomes: http://www.internationalgenome.org/
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
gnomAD Browser: http://gnomad.broadinstitute.org/
GraphPad: https://www.graphpad.com/
HGMD: http://www.hgmd.cf.ac.uk/ac/index.php
LOVD: https://databases.lovd.nl/shared/genes/NF1
Exome Variant Server: http://evs.gs.washington.edu/EVS/
OMIM: http://www.omim.org/
Change history
02 October 2018
A correction has been published to this Article. The PDF and HTML have been updated accordingly.
References
Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135:904–906.
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578.
Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81–88.
Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140–151.
Pinna V, Lanari V, Daniele P, et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015;23:1068–1071.
Rojnueangnit K, Xie J, Gomes A, et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat. 2015;36:1052–1063.
Koczkowska M, Chen Y, Callens T, et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. Am J Hum Genet. 2018;102:69–87.
Ekvall S, Sjörs K, Jonzon A, Vihinen M, Annerén G, Bondeson ML. Novel association of neurofibromatosis type 1-causing mutations in families with neurofibromatosis-Noonan syndrome. Am J Med Genet A. 2014;164A:579–587.
Nyström AM, Ekvall S, Strömberg B, et al. A severe form of Noonan syndrome and autosomal dominant café-au-lait spots—evidence for different genetic origins. Acta Paediatr. 2009;98:693–698.
Santoro C, Maietta A, Giugliano T, et al. Arg(1809) substitution in neurofibromin: further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur J Hum Genet. 2015;23:1460–1461.
Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111:1355–1381.
Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26:704–711.
Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet. 1989;26:712–721.
Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–66.
Van Es S, North KN, McHugh K, De Silva M. MRI findings in children with neurofibromatosis type 1: a prospective study. Pediatr Radiol. 1996;26:478–487.
Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70:138–143.
Cnossen MH, de Goede-Bolder A, van den Broek KM, et al. A prospective 10 year follow up study of patients with neurofibromatosis type 1. Arch Dis Child. 1998;78:408–412.
McGaughran JM, Harris DI, Donnai D, et al. A clinical study of type 1 neurofibromatosis in north west England. J Med Genet. 1999;36:197–203.
Thakkar SD, Feigen U, Mautner VF. Spinal tumours in neurofibromatosis type 1: an MRI study of frequency, multiplicity and variety. Neuroradiology. 1999;41:625–629.
Lin AE, Birch PH, Korf BR, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet. 2000;95:108–117.
Blazo MA, Lewis RA, Chintagumpala MM, Frazier M, McCluggage C, Plon SE. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224–229.
Khosrotehrani K, Bastuji-Garin S, Riccardi VM, Birch P, Friedman JM, Wolkenstein P. Subcutaneous neurofibromas are associated with mortality in neurofibromatosis 1: a cohort study of 703 patients. Am J Med Genet A. 2005;132A:49–53.
Plotkin SR, Bredella MA, Cai W, et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012;7:e35711.
Blanchard G, Lafforgue MP, Lion-François L, et al. Systematic MRI in NF1 children under six years of age for the diagnosis of optic pathway gliomas. Study and outcome of a French cohort. Eur J Paediatr Neurol. 2016;20:275–281.
Quintáns B, Pardo J, Campos B, et al. Neurofibromatosis without neurofibromas: confirmation of a genotype-phenotype correlation and implications for genetic testing. Case Rep Neurol. 2011;3:86–90.
Burgoyne AM, De Siena M, Alkhuziem M, et al. Duodenal-jejunal flexure GI stromal tumor frequently heralds somatic NF1 and notch pathway mutations. JCO Precis Oncol. 2017;1:1–12.
Kehrer-Sawatzki H, Mautner VF, Cooper DN. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet. 2017;136:349–376.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Tonsgard JH, Kwak SM, Short MP, Dachman AH. CT imaging in adults with neurofibromatosis-1: frequent asymptomatic plexiform lesions. Neurology. 1998;50:1755–1760.
Waggoner DJ, Towbin J, Gottesman G, Gutmann DH. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet. 2000;92:132–135.
Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis. 2011;6:18.
Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314.
Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumours in NF1. Neurology. 2005;65:205–211.
McCaughan JA, Holloway SM, Davidson R, Lam WWK. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J Med Genet. 2007;44:463–466.
Brems H, Pasmant E, Van Minkelen R, et al. Review and update of SPRED1 mutations causing Legius syndrome. Hum Mutat. 2012;33:1538–1546.
Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101–116.
Fleming AJ, Chi SN. Brain tumors in children. Curr Probl Pediatr Adolesc Health Care. 2012;42:80–103.
Sellmer L, Farschtschi S, Marangoni M, et al. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J Rare Dis. 2017;12:34.
Korf BR. Malignancy in neurofibromatosis type 1. Oncologist. 2000;5:477–485.
Tadini G, Milani D, Menni F, Pezzani L, Sabatini C, Esposito S. Is it time to change the neurofibromatosis 1 diagnostic critieria? Eur J Intern Med. 2014;25:506–510.
Szafranski P, Schaaf CP, Person RE, et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat. 2010;31:840–850.
Hirata Y, Brems H, Suzuki M, et al. Interaction between a domain of the negative regulator of the Ras-ERK pathway, SPRED1 protein, and the GTPase-activating protein-related domain of neurofibromin is implicated in Legius syndrome and neurofibromatosis type 1. J Biol Chem. 2016;291:3124–3134.
Evans DGR, Salvador H, Chang VY, et al. Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin Cancer Res. 2017;23:e46–e53.
Acknowledgements
We thank the individuals and their families for participating in this study. This work was supported by the Children’s Tumor Foundation by the Isaac and Sadie Fuchs Genotype–Phenotype Study (to L.M.M.) and by internal funds from the Medical Genomics Laboratory at UAB. M.K. is also affiliated with the Department of Biology and Medical Genetics at the Medical University of Gdansk in Poland. We thank our collaborators: Gail Graham (Children’s Hospital of Eastern Ontario in Ottawa), Stephanie M. Morris (Washington University in St. Louis), Dmitriy M. Niyazov (Ochsner Clinic Foundation in New Orleans), Catherine Ward-Melver (Akron Children’s Hospital), Lawrence Wong (Kaiser Permanente South Bay Medical Center in Harbor City), Katie Farmer (Tallahassee Memorial HealthCare), Monisha Samanta Kisling (Children’s National Medical Center in Washington), and Taylor Warner (University of Iowa Hospitals and Clinics) for confirming the accuracy of clinical information originally provided for their individuals as well as Ender Karaca (UAB) for performing targeted NF1 testing for UAB-R1873 family.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
A correction has been made to the footnotes of Table 2 which contained mistakes in the paper that was published online from September 7th to 16th, 2018. These mistakes were not created by the authors in any way and corrections were implemented as of September 17th. We apologize for the inconvenience to the authors and their readers.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koczkowska, M., Callens, T., Gomes, A. et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype–phenotype correlation. Genet Med 21, 867–876 (2019). https://doi.org/10.1038/s41436-018-0269-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0269-0
Keywords
This article is cited by
-
Genotype–phenotype correlations of neurofibromatosis type 1: a cross-sectional study from a large Chinese cohort
Journal of Neurology (2024)
-
Analysis of 200 unrelated individuals with a constitutional NF1 deep intronic pathogenic variant reveals that variants flanking the alternatively spliced NF1 exon 31 [23a] cause a classical neurofibromatosis type 1 phenotype while altering predominantly NF1 isoform type II
Human Genetics (2023)
-
Neurofibromatosis type1, type 2, tuberous sclerosis and Von Hippel-Lindau disease
Child's Nervous System (2023)
-
Age-dependent oral manifestations of neurofibromatosis type 1: a case–control study
Orphanet Journal of Rare Diseases (2022)
-
Neurofibromin and suppression of tumorigenesis: beyond the GAP
Oncogene (2022)
