
Abstract
Purpose
Pathogenic variants in neuroblastoma-amplified sequence (NBAS) cause an autosomal recessive disorder with a wide range of symptoms affecting liver, skeletal system, and brain, among others. There is a continuously growing number of patients but a lack of systematic and quantitative analysis.
Methods
Individuals with biallelic variants in NBAS were recruited within an international, multicenter study, including novel and previously published patients. Clinical variables were analyzed with log-linear models and visualized by mosaic plots; facial profiles were investigated via DeepGestalt. The structure of the NBAS protein was predicted using computational methods.
Results
One hundred ten individuals from 97 families with biallelic pathogenic NBAS variants were identified, including 26 novel patients with 19 previously unreported variants, giving a total number of 86 variants. Protein modeling redefined the β-propeller domain of NBAS. Based on the localization of missense variants and in-frame deletions, three clinical subgroups arise that differ significantly regarding main clinical features and are directly related to the affected region of the NBAS protein: β-propeller (combined phenotype), Sec39 (infantile liver failure syndrome type 2/ILFS2), and C-terminal (short stature, optic atrophy, and Pelger–Huët anomaly/SOPH).
Conclusion
We define clinical subgroups of NBAS-associated disease that can guide patient management and point to domain-specific functions of NBAS.
Similar content being viewed by others


INTRODUCTION
In 2010, NBAS (neuroblastoma-amplified sequence, MIM 608025)–associated disease was described in Yakuts, an isolated population living in the far east of the Russian Federation, carrying a homozygous missense variant in NBAS (c.[5741G>A]; resulting in p.[Arg1914His]). Their clinical phenotype comprised short stature with facial dysmorphism, optic atrophy, and Pelger–Huët anomaly, leading to the coining of the term SOPH syndrome (MIM 614800).1 In 2015, the discovery of biallelic NBAS variants in patients with recurrent acute liver failure (RALF) linked NBAS to hepatic disease and revealed the occurrence of pathogenic NBAS variants in the Caucasian and US populations.2 As extrahepatic findings such as facial abnormalities or short stature were also present in some of these patients with a predominant hepatic phenotype, we hypothesized that NBAS variants may cause a disease spectrum between the phenotypic features of SOPH syndrome and isolated RALF (infantile liver failure syndrome type 2/ILFS2, MIM 616483).3,4
Within just the next three years, 44 patients from 35 families with NBAS-associated disease were published or reported at conferences.4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36 The growing number of known affected individuals continuously expanded the phenotypic spectrum. The organ systems mainly involved were liver, skeletal systems including growth, the nervous system including the eye, the integument, the immune system, and the musculature. Additionally, facial dysmorphism was also prevalent. Given the fact that levels of the NBAS protein were reduced in patients’ fibroblasts (refs. 2,10, authors’ unpublished data), NBAS-associated disease is often termed as NBAS deficiency.
The wide spectrum of clinical symptoms associated with NBAS variants points to crucial functions of the NBAS protein in biology. The protein functions as a component of an endoplasmic reticulum (ER) tethering complex involved in the retrograde Golgi–ER transport.2,3,37 New data suggest an additional role of NBAS at the ER exit site for the formation of large transport vesicles for bulky cargo (e.g., collagen) in the secretory pathway.38 Recently, variants in RINT1, which encodes for a direct interaction partner of NBAS in the same tethering complex, were shown to cause human disease with a striking clinical overlap to NBAS deficiency.39 This strongly supports the hypothesis that dysfunction of the tethering complex is causative for at least a part of the phenotypic spectrum of NBAS-associated disease. Moreover, the discovery of cholestasis, acute liver failure, and neurodegeneration (CALFAN) syndrome due to variants in SCYL1, another disorder of intracellular trafficking with clinical and functional overlap to NBAS, enforces the link between disturbed Golgi–ER transport and human disease.40 Apart from its role in intracellular transport, NBAS was identified to be a mediator of nonsense-mediated messenger RNA (mRNA) decay (NMD), especially modulating genes associated with protein trafficking and ER-coupled protein modifications,41 but the role of altered NMD in NBAS deficiency remains to be elucidated.
Exploring clinical disease patterns associated with NBAS variants and their correlation to genotypes provides great chances to improve our understanding of the function of NBAS. It is the nature of case reports and case series that there is a lack of systematic analysis and to date, there is no unified approach for the attribution of patients to the associated clinical syndromes or the nomenclature of these. In this study, we systematically and quantitatively analyze the genotype and clinical phenotype of all patients with pathogenic NBAS variants currently known to us, including all previously reported cases and 26 novel patients from 23 families with 19 novel variants.
MATERIALS AND METHODS
Study design, recruitment of patients, data acquisition, and definition of variables
Individuals were recruited within an international, multicenter study. Inclusion criteria were rare biallelic variants in NBAS (NM_015909.3) classified as pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines for the interpretation of sequence variants and/or NBAS protein or RNA analysis in fibroblasts.42 Patients were excluded from the quantitative analysis of the clinical phenotype, if they fulfilled one of the following criteria: (1) patients aged less than 12 months at last assessment (to reduce the effect of low age on a possibly evolving phenotype), (2) presymptomatic diagnosis due to family screening, (3) presence of severe comorbidities unrelated to NBAS variants, and (4) lack of individualized clinical data.
Recruitment was via one of the following options: (1) individuals diagnosed via exome sequencing at the Institute of Human Genetics of the Technical University of Munich or another collaborative center, (2) individuals followed by one of the coauthors, or (3) previously published patients.
For options 1 and 2, clinical data were retrieved via case report forms and stored within a disease-specific database located at the Center for Child and Adolescent Medicine of the University Hospital Heidelberg. This included current follow-up visits of previously reported individuals.3,4 If available and consented by the parents, portrait photos of the patients were collected. In case of previously published patients reported by other groups, data were obtained from these publications; in case of missing data for the quantitative analysis, authors were contacted and asked to share additional information (see Acknowledgements). Duplicate case reports were omitted. Missing data were not imputed.
To reach out to other centers with diagnosed but unpublished individuals with NBAS-associated disease, we posted a call to participate within this study on Metab-L, an electronic mailing list on inborn errors of metabolism, on 5 December 2018.43
For the identification of previously published patients, a comprehensive literature search was performed using PubMed and Google Scholar search engines and the 2015–2018 abstract books from genetic, metabolic, and endocrinological scientific societies (for details see Supplementary Information). For quantitative analysis of the clinical phenotype, the following variables were analyzed within this study: country of patient’s origin, sex, age at last assessment, and whether the patient was dead or alive at that point as well as age and cause of death. Additionally, clinical features of the main organ systems involved were scrutinized (see legend of Fig. 1, nomenclature according to Human Phenotype Ontology [HPO] terminology).

Localization of NBAS variants and characterization of the study cohort: geographical origin, affected organ systems, and age distribution. (a) Top: nonsense and splice site variants as well as deletions of one or more exons. Bottom: missense variants and in-frame deletions. (b) Geographical origin. Due to the founder effect within the Yakut population, currently most patients originate from Russia. (c) Distribution of affected organ systems. The following clinical items were analyzed: acute liver failure, continuously elevated transaminases (abnormality of the liver); short stature (abnormality of growth); reduced bone mineral density, delayed closure of fontanelles, abnormalities of the vertebral column or cervical instability (abnormality of the skeletal system); motor delay, optic atrophy, intellectual disability (abnormality of the nervous system); cutis laxa (abnormality of the integument); decreased circulating IgG, reduced natural killer cell count, Pelger–Huët anomaly (abnormality of the immune system); muscular hypotonia, skeletal muscle atrophy (abnormality of the musculature). (d) Age distribution at last visit.
For the evaluation regarding facial dysmorphism, available facial 2D profiles of patients were uploaded to the Face2Gene RESEARCH app (FDNA Inc., Boston, MA44) to generate composite photos allowing a score distribution comparing them with age and gender-matched controls.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent to participate in the study was obtained from all patients or their parents in case of minor patients, except for cases where patient data were retrieved from publications. Additional informed consent was obtained from all patients for whom identifying information is included in this article. The study was approved by the ethical committee of the Technical University Munich and the University Hospital Heidelberg. The cut-off date for data analysis was 7 May 2019.
Identification of NBAS variants and ex vivo studies in patient and control fibroblasts
NBAS variants were identified using exome sequencing; carrier status of parents was confirmed via trio-exome sequencing or Sanger sequencing (details available upon request). NBAS protein levels in patient fibroblasts were measured by western blotting as described previously.2,40
Tertiary structure predictions and sequence analysis
Prediction of the tertiary structure of the human NBAS protein (Uniprot ID: A2RRP1) was performed by the HHpred web server45 using the PDB70 target database; all other parameters were set to default. We considered the hit that had the best alignment with the query protein as the model. The IUPRED2A web server46 was used to predict regions of disorder and MAFFT web server47 was used for the alignment of protein sequences.
Statistics
Count data from frequency tables were analyzed with log-linear models in R software, a language for statistical analysis and graphics (https://www.r-project.org) and visualized by mosaic plots with Pearson residual-based shadings.48 The world map was generated with the R package rworldmap.49 The medians of two samples were compared with a Mann–Whitney U test. For statistical evaluation of facial abnormalities, the area under the curve (AUC) of the receiver operating characteristic (ROC) curve was measured and statistical significance using the P value with the two-sided population proportions test was computed to compare each group with the age-/gender-matched controls.
RESULTS
Study population
The literature search identified 28 publications and 7 abstracts on individuals with biallelic pathogenic NBAS variants, published between February 2008 and April 2019. Thirty-three of the publications and abstracts were relevant and provided sufficient data for this study, reporting a total of 88 patients from 81 families. Twenty-three patients from 21 families within this cohort, harboring 19 novel variants, have not been reported in the literature before and were identified by one of the coauthors; additionally, three patients, so far only reported at conferences, were included in this study with more detailed information on the phenotype. These 26 patients from 23 families are termed “novel patients” (Table 1). For the quantitative analysis of the clinical phenotype, two individuals (previously unreported) were excluded because age at last assessment was before 12 months of age (NBAS 50, NBAS 64). Furthermore, one individual was excluded because he was identified in a family history–triggered investigation and presented no symptoms other than Pelger–Huët anomaly (NBAS 71).34 A fourth (previously unreported) individual was excluded due to comorbidity unrelated to NBAS variants (NBAS 61, Williams–Beuren syndrome).
Genetics
One hundred and ten individuals from 97 unrelated families with biallelic pathogenic NBAS variants were identified, with a total number of 86 different variants: 40 missense variants, 13 frameshift variants, 13 splice site variants, 12 nonsense variants, 5 exon deletions, 2 in-frame deletions, and 1 intronic variant (see Fig. 1a). There are 66 genotypes, 13 of them homozygous. The most frequent genotype (30 families) is homozygosity for the missense variant c.(5741G>A) leading to the amino acid substitution p.(Arg1914His), which however has only been reported in the Yakut population.1 Apart from this genotype, no specific variant combination is present in more than two unrelated families, which indicates an extremely high genetic heterogeneity. Most patients are homozygous for a missense variant (n = 47) or compound heterozygous for a missense in combination with a nonsense variant (n = 32) (see Supplemental Table S1). For the novel variants, functional validation by western blotting was done when fibroblasts were available (n = 9, see Supplementary Table S2).
Phenotypic spectrum
Patients originate from all continents except South America (Fig. 1b). The main organ systems or processes involved were liver, growth, skeletal system, nervous system, integument, immune system, and musculature (Fig. 1c), but also facial dysmorphism (Fig. 2c) and abnormalities of the endocrine system (including diabetes mellitus type 1). For the analysis of age, survival, and follow-up time, the 33 individuals from Yakutia1 were excluded due to a lack of individualized data. In the remaining patients, median age at last assessment was 7.0 years (total range: 1.0–42 years; 639 cumulative patient years) (Fig. 1d). Sixty-three patients were alive at the time of this report while 10 individuals had died—of whom 9 passed away due to acute liver failure (ALF) (median age at death: 2.3 years, range: 1.5–3.9 years) and one during liver transplant procedure at the age of 15 years (NBAS 51). Six patients underwent liver transplantation between the age of 1 and 6 years (median 2.8 years; age at first transplantation procedure in case of several transplantations). Follow-up time after transplantation is 0–14 years (cumulative time 44 years); none of these patients presented with a liver crisis after transplantation. For detailed information on individual patients see Supplementary Table S1.

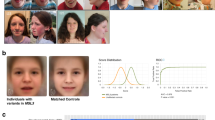
From protein to disease: tertiary structure of NBAS, localization of missense variants and in-frame deletions, disorder prediction, and phenotypic patterns of the three domain-related subgroups. (a) Tertiary structure modeling and disorder predictions of NBAS. Structural predictions were determined using the HHpred suite. The region 86–446 was modeled from the A chain of COMPASS histone methyltransferase in yeast (PDB: 6CHG_A; probability: 98.5; p value: 9.8e-12), and the region 722–1369 was modeled from the D chain of DslI transport complex in yeast (PDB: 3K8P_D; probability: 100; p value: 6.9e-66). Red inverted triangles indicate predicted regions with high disorder scores (1–41, 459–475, 644–657, 1382–1398, 1933–1947). The disorder score for the entire protein was determined using the IUPRED server. Missense variants and in-frame deletions are indicated as gray dots on the gene as well as gray Calpha spheres in the two modeled domains as best as possible. (b) Phenotypic patterns of the three subgroups β-propeller, Sec39 and C-terminal. (c) Facial phenotypes (composite faces). All patients tend to present thicker eyebrows and broader columella, but only patients attributed to the C-terminal group significantly differ from their controls, presenting triangular face, wide palpebral fissures, deep set eyes and thin upper lip vermilion, thick eyebrows, short philtrum, and small mouth. AUC area under the curve, ROC receiver operating characteristic.
Within the group of the 26 novel patients (Table 1) and as observed during follow-up of previously reported patients, new phenotypic aspects include inverted nipples at birth, lipodystrophy, joint laxity, dislocation of the lens, premature loss of milk teeth (connective tissue), relatively small foramen magnum, spinal cord compression with lower limb weakness (skeletal system), chronic lung disease with oxygen dependency and bilevel positive airway pressure (BiPAP) respiratory support, recurrent bacterial pneumonia (respiratory system) and elevated 3-methylglutaconic acid in urine during liver crises with normalization in the interval between crises (n = 2, metabolism).
Genotype–phenotype correlation
Given the heterogeneity of the individual patients’ phenotypes and the wide range of symptoms associated with biallelic pathogenic NBAS variants, we aimed to explore clinical subgroups. It has already been reported that patients with missense variants in the region coding for the Sec39 domain of NBAS have a predominant liver phenotype with RALF.3,35 Indeed, this holds true for patients compound heterozygous for a missense variant in the region coding for the Sec39 domain plus a loss-of-function variant, independent from its localization. Interestingly, there are no individuals with biallelic nonsense variants, except one with a late frameshift variant c.(6966_6969delinsTC) in the last exon in combination with the variant c.(5547delC) (NBAS 57). In this case, immunoblotting revealed a reduced protein concentration of NBAS with a protein band showing nearly full-length size, whereas there is no second band of smaller size. This argues for a truncated protein as a result to the c.(6966_6969delinsTC), p.(Gln2322Hisfs*18), whereas the transcript of c.(5547delC) likely undergoes NMD. In further immunoblots of patient cells harboring nonsense variants, no smaller band corresponding to the expression of a truncated protein has been observed (n = 12 [refs. 3,10] and authors’ unpublished data). These observations indicate that transcripts with nonsense variants undergo NMD independently from the location of the nonsense variant. Hence, we hypothesize, in line with the concept of edgetic perturbations of protein functions,50 that the localization of the missense variants or in-frame deletions determines the phenotype of the respective patient.
To test this hypothesis, we first grouped missense variants and in-frame deletions according to their localization, whereas the latter is defined as the deletion of one amino acid. To provide insights into the structural location of each variant, we predicted structures using HHpred,45 which identified a single significant hit for the region 722–1369 and several significant hits in the region 86–446. The remaining part of the protein displayed no significant hits. Secondary structure45,51 and disorder52 predictions suggest several discrete regions in human NBAS (Uniprot ID: A2RRP1): a partly disordered, partly helical N-terminus (residues 1–85), a region consisting of mostly β-strands in WD40 repeats (86–446), with the remainder of the protein being predominantly helical (447–2371). Prediction of tertiary structure using HHpred web server45 defined an N-terminal β-propeller region roughly from residues 86 to 446, and an α-solenoid (TPR repeat) regions at residues 722–1369. IUpred2 suggests short regions of disorder that partly delineate these structures (Fig. 2a). Our defined β-propeller region is considerably (approximately 80 amino acids) longer than defined previously,2 where definition was based on a weak match to a domain from a quinoprotein amine dehydrogenase (InterPro: IPR011044). Our match is highly significant and agrees well with the predictions of β-strands, which are largely absent in the other parts of the protein. We found only one significant α-solenoid structure match, which was to Sec39 from yeast,53 which is the only (to date) structure of a Sec39 domain containing protein. The alignment covers the entirety of this structure, but only a small fraction of the NBAS C-terminus. This fact, plus the tendency of long-predicted helices, argues that likely we are not detecting long segments of helical repeat structures, since they are highly variable in sequence.
Interestingly, most missense variants and in-frame deletions cluster in the coding regions of the β-propeller domain, the Sec39 domain, and the C-terminal part of NBAS as well as in proximity to the regions of disorder identified by IUPred2 (Figs. 1a, 2a). Furthermore, we found these mutated residues more evolutionarily conserved than other residues (MAFFT47, Mann–Whitney U test; p < 0.05). Loss-of-function (nonsense or splice site) variants are found more evenly distributed. Seven patients are biallelic for a nonsense plus a splice site variant or two splice site variants. For six of these seven patients (NBAS 6, 11, 47, 48, 79, 80) with four different splice site variants (c.[6237-3C>G]; c.[6432+1_6433-1]_[6711 + 1_6712-1]del; c.[6840G>A]; c.[6840G>T]), immunoblotting and/or complementary DNA (cDNA) analysis revealed that mutant RNA escapes NMD leading to the expression of a shorter protein (refs. 3,24,35 and authorsʼ data, available upon request). For the seventh patient (NBAS 49), no fibroblasts were available.26 Accordingly, these variants were considered distinct to loss-of-function variants and—together with all missense variants and in-frame deletions—called determining variants in the following. Based on the localization of the determining variants, patients were attributed to three groups: β-propeller, Sec39, and C-terminal. Strikingly, these three groups have different clinical patterns; whereas determining variants in the coding region for the Sec39 domain are mainly associated with ALF, determining variants in the coding region for the C-terminus show a predominant multisystemic phenotype. Patients with determining variants in the region coding for the β-propeller domain, however, present with a combined severe phenotype including both ALF and multisystemic features (Fig. 2b).
Four patients are compound heterozygous for missense variants in regions coding for different domains and can therefore not be clearly attributed to one of the three groups based on their genotype. Their clinical presentation includes features of the respective subgroups, underlining a close relationship between variant localization and clinical phenotype (see Supplementary Table S1). The six patients with confirmed aberrant protein or transcript due to a splice site variant were attributed according to the localization of these splice site variants, whereas patient NBAS 49 was not attributed to a group.
To test whether differences in phenotypes between the three groups are statistically significant, log-linear models were calculated. While there was no difference of prevalence of NK cell deficiency and intellectual disability, all other features tested distribute unequally (Fig. 3 and Supplementary Fig. 1). Short stature was present in all three groups, but height standard deviation score (SDS) differs significantly between Sec39 and C-terminal (Wilcoxon rank sum test p = 0.00034, Fig. 3).

Subgroups differ significantly regarding key clinical features. Clinical features of all affected organ systems differ significantly between the three subgroups β-propeller, Sec39, and C-terminal. Gray mosaics indicate Pearson residuals between 1.96 and −1.96 (equal distribution), blue indicates more observed cases than expected, and red indicates fewer observed cases than expected from marginal frequencies (see Pearson scale). Age distribution within the three groups: β-propeller (median: 8.5 years, standard deviation: 6.16), Sec39 (median: 8.0 years, standard deviation: 9.09), and C-terminal (median 6.0 years, standard deviation: 5.69). SDS, standard deviation score.
For the analysis of facial abnormalities, photographs from 39 patients were available (refs. 1,3,4,6,8,10,11,14,17,24,35,54,55 and authorsʼ patients) and assigned to one of the three groups (β-propeller n = 10, Sec39 n = 11, C-terminal n = 18). There is a significant difference in the facial presentation of the C-terminal group compared with the matched control cohort (p = 0.009), while the other two groups (β-propeller and Sec39) did not differ significantly from their controls (Fig. 2c).
DISCUSSION
We present an international, multicenter cross-sectional study on the clinical phenotypes of individuals with NBAS-associated disease in a cohort of 110 patients from 97 families with a total of 639 patient years. This report adds data of 26 novel patients to the literature, including 19 previously unreported variants, creating a total number of 86 known pathogenic variants. Genotype–phenotype associations are explored based on variant localizations including models of the tertiary structure of NBAS.
NBAS-associated disease is related to a wide spectrum of clinical signs and symptoms mainly affecting liver, growth, integument, immune system, skeletal system, nervous system, and musculature. In line with the concept of edgetic perturbation,50 our study demonstrates that phenotypes can be grouped into three clinical subgroups based on the localization of missense variants or in-frame deletions to one of the two regions coding for the known domains or the C-terminal part of NBAS. These three groups differ significantly. Considering the currently used terms for clinical syndromes associated with pathogenic NBAS variants, ILFS2 (MIM 616483) resembles patients with variants affecting Sec39, whereas SOPH syndrome (MIM 614800) resembles patients with variants affecting the C-terminal part of the gene. Interestingly, patients with variants affecting the β-propeller domain present a combined phenotype that may be considered as an overlap of ILFS2 and SOPH syndrome, including clinical features of both groups.
These findings have a direct impact on patient management, e.g., regular screening for reduced IgG levels or optic atrophy for patients with variants affecting the β-propeller domain or the C-terminus and high awareness for the risk of fever-related, life-threatening ALF in patients with variants affecting the β-propeller or the Sec39 domain. The latter is of special importance, as early and aggressive antipyretic treatment together with glucose and lipid infusion during ALF has been reported to reduce severity of ALF in NBAS deficiency.3,14
A recent study investigated the facial characteristics of patients with NBAS-associated disease using DeepGestalt,35 confirming the previously reported characteristics of hypotelorism, thin lips, pointed chin, and progeroid appearance.3 However, facial phenotypes had not been related to the remaining symptoms or genotypes of the patients. We demonstrate that the facial phenotype depends on the genotype (Fig. 2c).
Given the rapidly increasing patient numbers since its first report, NBAS-associated disease is likely still an underdiagnosed condition. As there are no biomarkers to date, diagnosis relies on genetic testing, mainly exome sequencing. The definition of clinical subgroups with distinct phenotypic patterns will help to identify clinical candidates, facilitates phenotype-based variant filters in the analysis of exome data, and will help to establish earlier diagnosis in patients with NBAS-associated disease.
Our study is limited by the fact that the majority of study individuals are still in infancy or early childhood. Aspects of an evolving clinical phenotype might thus be underrepresented suggesting additional follow-up investigations are needed, including the reevaluation of the described clinical patterns within the growing patient numbers. In addition, our data also suggest that studies not only on natural history but also pathomechanism in NBAS deficiency should differentiate between the different clinical subtypes of the disease, based on the genotype. Apart from patient management, our findings are of importance for the understanding of the protein function, suggesting domain-specific roles. Overall, NBAS resembles several proteins in the nuclear pore, clathrin-coated vesicles, and intraflagellar transport, many of which also contain an N-terminal β-propeller (WD repeat) region with a flexible, long C-terminal region consisting of ɑ-solenoid (TPR) repeats.56 Generally, these proteins serve as scaffolds to interact with multiple protein partners. Though we could not identify any sequence-similar templates to model an interaction,57 it is very likely that both the propeller and solenoid regions help binding to one or more of the NBAS interaction partners. β-propellers have a preferred binding site at the top of the barrel,58 though as scaffolds they can, in principle, bind at alternative sites. The apparent accumulation of variants on one face suggests that they could be affecting the same (albeit unknown) set of interaction partners binding to this side of the propeller.
Genotype–phenotype associations argue for a role of the Sec39 domain for liver function, thereby associating liver dysfunction with altered intracellular transport, as Sec39 is required for tethering at the ER in the syntaxin18 complex. This is underlined by the fact that deficient RINT1, a direct interaction partner of NBAS within this complex, also leads to RALF. ER stress and altered autophagy may be involved in the pathomechanism,2,39 but mechanisms of fever-dependent, syntaxin18–related RALF are still poorly understood.
The high prevalence of various multisystemic symptoms associated with variants affecting the C-terminal part of NBAS point to important functions of this part of the protein, which so far has no defined domains and for which no reliable 3D modeling is currently possible. It is an interesting observation that the C-terminal missense variants cluster in close proximity to the variant prevalent in the Yakut population c.(5741G>A) and in close relation to a highly disordered region (IUPred2 score), which might suggest a short peptide binding region (or linear motif).59
The combined severe phenotype caused by variants in the coding region of the β-propeller domain, a domain that is per se difficult to fold,60 might reflect disturbed functions of both the Sec39 and C-terminal parts due to an altered β-propeller. This might be due to hampered protein interaction with other partners or misfolding of the whole protein. The latter could be of interest for a therapeutic approach to promote folding by specific chaperones in the future. However, further studies are needed to improve our knowledge about the tertiary structure of NBAS, domain-specific protein functions, and potential sites of interaction.
This is the first work systematically and quantitatively analyzing patients with NBAS-associated disease, based on the largest cohort studied so far, including more than 100 patients and adding 26 novel patients to the literature. Based on the localization of missense variants or in-frame deletions, most affected individuals can be grouped into three subgroups. We suggest to name the distinct clinical subgroups ILFS2 (Sec39), SOPH (C-terminal), and ILFS2-SOPH (β-propeller). Our study facilitates patient management, which should be guided by the specific phenotypic patterns. Ultimately, improved knowledge of domain-specific functions will boost our understanding of both biology and human disease related to variants in NBAS.
References
Maksimova N, Hara K, Nikolaeva I, et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Huet anomaly. J Med Genet. 2010;47:538–548.
Haack TB, Staufner C, Kopke MG, et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy. Am J Hum Genet. 2015;97:163–169.
Staufner C, Haack TB, Kopke MG, et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts. J Inherit Metab Dis. 2016;39:3–16.
Kortum F, Marquardt I, Alawi M, et al. Acute liver failure meets SOPH syndrome: a case report on an intermediate phenotype. Pediatrics. 2017;139:e20160550.
Segarra NG, Ballhausen D, Crawford H, et al. NBAS mutations cause a multisystem disorder involving bone, connective tissue, liver, immune system, and retina. Am J Hum Genet A. 2015;167A:2902–2912.
Capo-Chichi JM, Mehawej C, Delague V, et al. Neuroblastoma Amplified Sequence (NBAS) mutation in recurrent acute liver failure: confirmatory report in a sibship with very early onset, osteoporosis and developmental delay. Eur J Med Genet. 2015;58:637–641.
Buote C Application clinique du séquençage de l’exome pour le diagnostic moléculaire des syndromes polymalformatifs, Sherbrooke, CA: Department of Biochemistry, University of Sherbrooke; 2015.
Haskins-Olney A. NBAS-related SOPH syndrome with immunodeficiency in a North American patient. Paper presented at: ACMG Annual Clinical Genetics Meeting; Tampa, FL; March 8–12, 2016.
Rothenbuhler A. NBAS mutations, a new monogenic cause of DISOPHAL, a new syndrome with type 1 diabetes (T1D). Paper presented at: 55th Annual European Society for Paediatric Endocrinology Meeting; Paris; September 10–12, 2016.
Balasubramanian M, Hurst J, Brown S, et al. Compound heterozygous variants in NBAS as a cause of atypical osteogenesis imperfecta. Bone. 2017;94:65–74.
Regateiro FS, Belkaya S, Neves N, et al. Recurrent elevated liver transaminases and acute liver failure in two siblings with novel bi-allelic mutations of NBAS. Eur J Med Genet. 2017;60:426–432.
Calvo PL, Tandoi F, Haak TB, et al. NBAS mutations cause acute liver failure: when acetaminophen is not a culprit. Ital J Pediatr. 2017;43:88.
Hasosah MY, Iskandarani AI, Shawli AI, Alsahafi AF, Sukkar GA, Qurashi MA. Neuroblastoma amplified sequence gene mutation: a rare cause of recurrent liver failure in children. Saudi J Gastroenterol. 2017;23:206–208.
Cardenas V, DiPaola F, Adams SD, Holtz AM, Ahmad A. Acute liver failure secondary to neuroblastoma amplified sequence deficiency. J Pediatr. 2017;186:179–182.
Li JQ, Qiu YL, Gong JY, et al. Novel NBAS mutations and fever-related recurrent acute liver failure in Chinese children: a retrospective study. BMC Gastroenterol. 2017;17:77.
Park JW, Lee SJ. Foveal hypoplasia in short stature with optic atrophy and Pelger-Huet anomaly syndrome with neuroblastoma-amplified sequence (NBAS) gene mutation. J AAPOS. 2017;22:334.
Kim YM, Lee YJ, Park JH, et al. High diagnostic yield of clinically unidentifiable syndromic growth disorders by targeted exome sequencing. Clin Genet. 2017;92:594–605.
He TY, Zhang N, Xia Y, Luo Y, Li CR, Yang J. [Short stature, optic nerve atrophy and Pelger-Huët anomaly syndrome with antibody immunodeficiency and aplastic anemia: a case report and literature review]. Zhonghua Er Ke Za Zhi. 2017;55:942–946.
Yao Q, Wu W, Luo X. SOPH syndrome causes recurrent acute liver failure with onset in infancy. Paper presented at: 13th International Congress of Inborn Errors of Metabolism (ICIEM); Rio de Janeiro; September 5–8, 2017.
Mungan N, Yildizdas D, Arikan C, et al. Recurrent acute liver failure in a family with NBAS gene mutation and successful liver transplantation: first cases from Turkey. Journal of Inborn Errors of Metabolism & Screening. 2017;5:399.
Yadav A, Hopkin RJ, Schorry EK. SOPH syndrome: multisystem disorder with facial dysmorphism, skeletal dysplasia, episodic liver failure, immune dysfunction and intellectual disability. Paper presented at: American Society of Human Genetics Meeting; Orlando, FL; October 17–21, 2017.
Wang J, Pu Z, Lu Z. Targeted nextgeneration sequencing reveals two novel mutations of NBAS in a patient with infantile liver failure syndrome 2. Mol Med Rep. 2018;17:2245–2250.
Sunwoo Y, Kim YM, Kim EN, Oh SH, Lee BH. Severe form of neuroblastoma amplified sequence deficiency in an infant with recurrent acute liver failure. Pediatr Int. 2018;60:302–304.
Palagano E, Zuccarini G, Prontera P, et al. Mutations in the neuroblastoma amplified sequence gene in a family affected by acrofrontofacionasal dysostosis type 1. Bone. 2018;114:125–136.
Li X, Cheng Q, Li N, et al. SOPH syndrome with growth hormone deficiency, normal bone age, and novel compound heterozygous mutations in NBAS. Fetal Pediatr Pathol. 2018;37:404–410.
Godinez-Zamora GF, Baeza-Capetillo P, Villaseñor-Dominguez A, et al. A novel mutation in NBAS causes SOPH syndrome. Paper presented at: American Society for Human Genetics Annual Meeting; San Diego, CA; October 16–20, 2018.
Konstantopoulou VK, Moeslinger DM, Goeschl BG, Roscher A. Severe hypoglycemia and seizures as the first signs in NBAS deficiency. J Inherit Metab Dis. 2018;41 (Suppl 1):S37–S219.
Thuriot F, Buote C, Gravel E, et al. Clinical validity of phenotype-driven analysis software PhenoVar as a diagnostic aid for clinical geneticists in the interpretation of whole-exome sequencing data. Genet Med. 2018;20:942–949.
Mallakmir S, Nagral A, Bagde A, Mirza D, Merchant R, Yewale V. Mutation in the neuroblastoma amplified sequence gene as a cause of recurrent acute liver failure, acute kidney injury, and status epilepticus. J Clin Exp Hepatol (in press).
Ranucci G, Lenz D, Staufner C, et al. Fever induced recurrent acute liver failure as a dominant feature in congenital disorders of intracellular trafficking. J Inherit Metab Dis. 2018;41 (Suppl 1):S37–S219.
Giatropoulou S, König R, Wudy SA, et al. NBAS gene mutation causes insulin-dependent diabetes mellitus in a patient with a multisystem disorder consisting immunodeficiency and extremely short stature. Paper presented at: 57th Annual Meeting of the European Society for Paediatric Endocrinology; Athens, Greece; September 27–29, 2018.
Rius R, Riley LG, Guo Y, et al. Cryptic intronic NBAS variant reveals the genetic basis of recurrent liver failure in a child. Mol Genet Metab. 2019;126:77–82.
Nucci F, Lembo A, Farronato M, Farronato G, Nucci P, Serafino M. Oculofacial alterations in NBAS-SOPH like mutations: case report. Eur J Ophthalmol. 2019;8:1120672119836335.
Ono S, Matsuda J, Watanabe E, et al. Novel neuroblastoma amplified sequence (NBAS) mutations in a Japanese boy with fever-triggered recurrent acute liver failure. Hum Genome Var. 2019;6:2.
Carli D, Giorgio E, Pantaleoni F, et al. NBAS pathogenic variants: defining the associated clinical and facial phenotype and genotype–phenotype correlations. Hum Mutat. 2019;40:721–728.
Fischer-Zirnsak B, Koenig R, Alisch F, et al. SOPH syndrome in three affected individuals showing similarities with progeroid cutis laxa conditions in early infancy. J Hum Genet. 2019;64:609–616.
Aoki T, Ichimura S, Itoh A, et al. Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 complex implicated in Golgi-to-endoplasmic reticulum retrograde transport. Mol Biol Cell. 2009;20:2639–2649.
Raote I, Ortega-Bellido M, Santos AJ, et al. TANGO1 builds a machine for collagen export by recruiting and spatially organizing COPII, tethers and membranes. eLife. 2018;7:e32723.
Cousin MA, Conboy E, Wang JS, et al. RINT1 bi-allelic variations cause infantile-onset recurrent acute liver failure and skeletal abnormalities. Am J Hum Genet. 2019;105:108–121.
Lenz D, McClean P, Kansu A, et al. SCYL1 variants cause a syndrome with low gamma-glutamyl-transferase cholestasis, acute liver failure, and neurodegeneration (CALFAN). Genet Med. 2018;20:1255–1265.
Longman D, Hug N, Keith M, et al. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013;41:8319–8331.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Renner C, Razeghi S, Richter T, Ueberall MA, Schless A. Metab-L: an electronic mailing list on inborn errors of metabolism. Acta Paediatr. 1997;86:892–894.
Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–64.
Zimmermann L, Stephens A, Nam SZ. et al. A completely reimplemented MPI Bioinformatics Toolkit with a nw HHpred server at its core. J Mol Biol. 2018;430:2237–2243.
Meszaros B, Erdos G, Dosztanyi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46(W1):W329–W337.
Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47(W1):W5–W10.
Meyer D, Zeileis A, Hornik K. The Strucplot Framework: Visualizing Multi-way Contingency Tables with vcd. J Stat Softw. 2006;17:48.
South A. rworldmap: a new R package for mapping global data. R J. 2011;3:35–43.
Zhong Q, Simonis N, Li QR, et al. Edgetic perturbation models of human inherited disorders. Mol Syst Biol. 2009;5:321.
Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43(W1):W389–W394.
Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434.
Ren Y, Yip CK, Tripathi A, et al. A structure-based mechanism for vesicle capture by the multisubunit tethering complex Dsl1. Cell. 2009;139:1119–1129.
Megarbane A, Samaras L, Chedid R, et al. Developmental delay, dysmorphic features, neonatal spontaneous fractures, wrinkled skin, and hepatic failure: a new metabolic syndrome? Am J Hum Genet A. 2008;146A:3198–3201.
Prontera P, Urciuoli R, Siliquini S, et al. Acrofrontofacionasal dysostosis 1 in two sisters of Indian origin. Am J Hum Genet A. 2011;155a:3125–3127.
van Dam TJ, Townsend MJ, Turk M, et al. Evolution of modular intraflagellar transport from a coatomer-like progenitor. Proc Natl Acad Sci U S A. 2013;110:6943–6948.
Aloy P, Russell RB. Interrogating protein interaction networks through structural biology. Proc Natl Acad Sci U S A. 2002;99:5896–5901.
Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci. 2010;35:565–574.
Gouw M, Michael S, Samano-Sanchez H, et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018;46(D1):D428–D434.
Willison KR. The substrate specificity of eukaryotic cytosolic chaperonin CCT. Philos Trans R Soc Lond B Biol Sci. 2018;373:20170192.
Acknowledgements
We thank Yoo-Mi Kim for providing additional, updated clinical information on patients NBAS 42 and NBAS 43 and John Christodoulou and Shanti Balasubramaniam for providing additional, updated clinical information on patient NBAS 63. Anne Davit Spraul is acknowledged for genetic diagnosis of patients NBAS 46 and NBAS 82, Oanez Ackermann and Dalila Habes are acknowledged for follow-up and Philippe Yakonowsky for his contribution to data collection of these patients. We thank Selina Wächter and Caterina Terrile for excellent technical assistance and Katharina Mayerhanser for organizational support. C.S. is supported by the Dietmar Hopp Foundation, St. Leon-Rot, Germany (grant number 23011235). D.L. is supported by the Deutsche Leberstiftung (grant number S163/10052/2018). J.A.C. is supported in part by the Jeffrey Model Foundation. R.B.R. and G.D.D. are supported by the Wellcome Trust grant 210585/B/18/Z (Impact of missense mutations in recessive Mendelian disease: insight from ciliopathies).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Staufner, C., Peters, B., Wagner, M. et al. Defining clinical subgroups and genotype–phenotype correlations in NBAS-associated disease across 110 patients. Genet Med 22, 610–621 (2020). https://doi.org/10.1038/s41436-019-0698-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0698-4
Keywords
This article is cited by
-
A novel variant in NBAS identified from an infant with fever-triggered recurrent acute liver failure disrupts the function of the gene
Human Genome Variation (2023)
-
NBAS, a gene involved in cytotoxic degranulation, is recurrently mutated in pediatric hemophagocytic lymphohistiocytosis
Journal of Hematology & Oncology (2022)
-
NBAS deficiency due to biallelic c.2809C > G variant presenting with recurrent acute liver failure with severe hyperammonemia, acquired microcephaly and progressive brain atrophy
Metabolic Brain Disease (2021)
-
Molekulare Medizin: Pathobiochemie als Schlüssel zur personalisierten Therapie vererbter Krankheiten
Monatsschrift Kinderheilkunde (2021)
-
NBAS Variants Are Associated with Quantitative and Qualitative NK and B Cell Deficiency
Journal of Clinical Immunology (2021)
