
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects motor neurons, causing progressive muscle weakness and respiratory failure. The presence of an expanded hexanucleotide repeat in chromosome 9 open reading frame 72 (C9ORF72) is the most frequent mutation causing familial ALS and frontotemporal dementia (FTD). To determine if suppressing expression of C9ORF72 gene products can reduce toxicity, we designed a set of artificial microRNAs (amiRNA) targeting the human C9ORF72 gene. Here we report that an AAV9-mediated amiRNA significantly suppresses expression of the C9ORF72 mRNA, protein, and toxic dipeptide repeat proteins generated by the expanded repeat in the brain and spinal cord of C9ORF72 transgenic mice.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis is a lethal neurodegenerative disease that affects approximately 2 in every 100,000 people worldwide [1]. ALS patients develop progressive muscle wasting and respiratory failure and have an average life expectancy of 2–5 years after symptom onset [2]. A hexanucleotide repeat expansion (HRE) consisting of GGGGCCn in intron 1 of the Chromosome 9 Open Reading Frame 72 (C9ORF72) gene is the most common cause of both inherited (40%) and sporadic (5–6%) ALS [3,4,5] as well as the most common genetic cause of frontotemporal dementia (FTD) [6]. Healthy individuals typically harbor 2–24 repeats, while patients with C9ORF72 ALS harbor hundreds or even thousands of the repeats [7, 8]. While the repeat length varies in number in different tissues, the most extensive expansions observed to date are in the brain [3, 5].
Three main splice variants have been validated by NCBI as the major products of the C9ORF72 gene: Variant 1 (NM_145005.5), Variant 2 (NM_018325.3), and Variant 3 (NM_001256054.1). Variants 1 and 3 contain the hexanucleotide expansion and are the least abundant; by contrast, the most abundant variant, V2, does not encompass the expansion. C9ORF72 is involved in multiple processes, including endosomal trafficking, nucleocytoplasmic transport, autophagy induction, and lysosomal biogenesis in diverse cell types, including motor neurons [9, 10]. The HRE appears to exert diverse adverse effects.
The HRE gives rise to three pathological hallmarks of C9ORF72 ALS: (1) haploinsufficiency, with reduced levels of mRNA leading to compromised neuronal viability; [10, 11] (2) nuclear accumulation of sense and antisense transcripts of the C9ORF72 HRE that sequester RNA binding and/or splicing proteins, forming RNA foci; [3, 12, 13] (3) repeat-associated non-ATG (RAN) translation of sense and antisense transcripts generating toxic dipeptide repeat proteins (DPRs) glycine-alanine (GA), glycine-proline (GP), and glycine-arginine (GR) DPRs in the sense direction and GP, proline-alanine (PA), and proline-arginine (PR) in the antisense direction [13, 14]. These aggregation-prone DPRs are found in the CNS and cerebrospinal fluid of C9ORF72 ALS and FTD patients [14,15,16] and are toxic in cell culture [17,18,19] and in animal models [20,21,22]. Although the full mechanisms by which the HRE causes motor neuron death are unknown, studies suggest a prominent role for the two gain of function pathologies, toxic DPRs and RNA foci. Haploinsufficiency of C9ORF72 by itself does not lead to motor neuron death as C9orf72 knockout mice do not show a motor phenotype. It is likely that there is evidence for a cooperative pathogenesis between gain- and loss-of-function mechanisms, in which C9ORF72 haploinsufficiency impairs clearance of DPRs, making motor neurons hypersensitive to DPR pathology [10].
Currently, treatment options in ALS are restricted to three FDA-approved medications that demonstrate only modest clinical efficacy (riluzole [23], edaravone [24, 25] and Relyvrio [26]). To develop an effective treatment for C9ORF72-mediated ALS, we have explored the use of RNA interference (RNAi) to achieve post-transcriptional suppression of the C9ORF72 gene.
We hypothesized that adeno-associated virus (AAV) administered via the cerebrospinal fluid will deliver the artificial microRNA (amiR) targeting C9ORF72 transcripts to cells in the central nervous system, and that the amiR will be processed in the nucleus and exported to the cytoplasm to suppress C9ORF72 transcripts. Silencing of C9ORF72 in this manner should reduce cytoplasmic levels of C9ORF72 transcripts, including transcripts carrying the disease-causing repeat expansion. While the amiR, which is exported to the cytoplasm, is not expected to shuttle back to the nucleus and affect RNA foci, it is expected to depress levels of cytoplasmic C9ORF72 transcripts containing the HRE, thereby ameliorating extra-nuclear toxicity from DPRs.
Methods
amiRNA design and cloning
All amiRs targeting C9ORF72 (Table 1) were cloned into the miR-155 backbone and then cloned into two entry GFP-expressing plasmids with different promoters: (1) the chicken beta actin (CBA) promoter with a CMV enhancer, and (2) the H1 promoter. The H1-amiRC9 and control GFP constructs were packaged into rAAV9 vectors.
Cell culture
For plasmid screening, HEK293T cells were seeded (1.2 E5 cell/ml) in a 24 well plate, maintained with DMEM media (11995-073; Gibco), 10% Fetal Bovine Serum FBS (F-0926, Sigma-Aldrich) and 1% penicillin/streptomycin (30-001-CI, Corning). The cells were transfected after 24 h using jetPRIME Reagent (Polyplus) and 1 µg of each plasmid DNA (CB-GFP, CB-GFP-amiRs) (Table 1). Forty-eight hours post transfection, GFP expression was visually assessed; cells PBS washed and collected using Trizol Reagent (Gibco BRL, Life Technologies, USA) for RNA isolation.
For primary cortical neuron experiments, homozygous BAC112 mice (JAX Stock# 023099, previously described by O’Rourke [27]) were crossed with female WT C57BL/6 mice to generate heterozygous embryos. Cortical cells were dissected at embryonic day 15 and pooled from multiple embryos. Mouse cortical cells were seeded at 50,000 cells/cm2 in six well plates treated with poly-d-lysine (P4707; Sigma-Aldrich) and four-well chamber slides (154917; Thermo Scientific). Cells were maintained with Neurobasal media (21103049; Invitrogen), Glutamax (35050-061; Invitrogen), 2% penicillin–streptomycin (15110122; Invitrogen) and B27 supplement (17504-001; Invitrogen). At day in vitro four (DIV4) cells were infected with rAAV9-CB-GFP and rAAV9-H1-amiRC9-CB-GFP at an MOI of 50,000. At DIV10, cells were rinsed with PBS and collected for RNA or protein isolation or for RNA foci analysis.
Animal experiments
All mouse experiments were conducted at UMASS Medical School following strict protocols approved by the Institutional Review Board. For animal studies we used heterozygous BAC112 mice. Homozygous male BAC112 mice were crossed with female WT C57BL/6 mice to generate heterozygous mice.
Bilateral striatal injections
4–6 months old heterozygous BAC112 mice were gender and transgene copy number matched, into three groups: PBS, rAAV9-CB-GFP, rAAV9-H1-amiRC9-CB-GFP. Mice were anesthetized with an intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg) injection. Mice were positioned in a stereotaxic frame and a sterile scalpel was used to cut through the skin. The skull was drilled at positions AP + 1, ML +-2, DV-2.5, following the mouse Stereotaxic Atlas coordinates for the striatum. The mice were injected at a 0.5 nl/ml flow with a volume of 5 µl per side, at a final dose of 1E11 genomic copies (GC) per mouse. The mice were PBS cardiac perfused at 16 weeks post injection and tissues were harvested. To harvest the injected striata, we used the 1MM mouse matrix (CP68-1175-1 SouthPoint surgical supply) taking sections 2–6 of the brain. A GFP positive 2 mm punch encompassing the injection site, was extracted and flash frozen in liquid nitrogen for further RNA and protein downstream processing.
ICV injections in neonates
Postnatal day 1 pups were separated from their mother and individually anesthetized with isoflurane and placed on a heated pad. The pups were injected using a 10 µl Hamilton Syringe with Removable Needle (55750-01: Hamilton) with 2 µl of vector into the lateral ventricle of each hemisphere, 2 mm from the transverse sinus and 1 mm to either side of the superior sagittal sinus, to a depth of 2 mm. Afterwards, they were placed on a heating pad until they regained consciousness. The pups were then rubbed against the bedding and placed back with the mother. Multiple litters were injected with the vectors: AAV9-CB-GFP 5E12 GC/ml; AAV9-H1-amiRC9-CB-GFP 5E12 GC/ml. Animals were perfused using PBS 16 weeks post injection. The whole brain and spinal cord were extracted, and the frontal lobe was dissected. The left side of the frontal lobe was flash frozen, and the right side was embedded in OCT for cryo-sectioning and staining.
Temporal vein injections in neonates
Postnatal day 1 pups were separated from their mother and individually anesthetized with isoflurane and placed on a heated pad. A Hamilton Syringe was used to inject 50 µl of vector at a final dose of 2E11 GC. We used the same vectors as described for the ICV treatment. After injection the pups were rubbed against their bedding to prevent rejection from the mother and placed back in the cage. Mice were euthanized 16 weeks post injection following the same collection procedure as for the ICV samples.
Laser capture microdissection of motor neurons
Flash-frozen cervical cords were cryo-sectioned into 7-µm-thick sections and collected on VWR VistaVision™ HistoBond® Adhesive Microscope Slides (Germany). Sections were first treated with RNAlater™-ICE (AM7030, Invitrogen, USA), which was followed by staining using a Histogene™ LCM Frozen Section Staining Kit (KIT0401, Applied biosystems). Then sections were fixed and dehydrated with 70-95-100% ethanol followed by xylene. LCM was then performed using the ArcturusXT™ Laser Capture Microdissection system (ThermoFisher, Waltham, MA, USA). The motor neurons were captured on CapSure™ Macro LCM Caps (LCM0211, ThermoFisher, Waltham, MA, USA). Three caps per animal were collected with approximately ~200 ventral horn motor neurons per cap.
RNA extraction, cDNA synthesis, DNA extraction and ddPCR
Frozen tissue samples were homogenized in a gentleMACS Dissociator (Miltenyi Biotec, USA) before total RNA extraction with Trizol (Life Technologies, USA) following manufactures protocol. For RNA extraction of cells, samples were collected in Trizol reagent (Gibco, Life Technologies), the aqueous phase was added to a Zymo column for RNA purification following the Zymo Direct-zol (Zymo research R2050) extraction protocol. RNA extraction of LCM isolated motor neurons was executed using the PicoPure RNA extraction kit following manufactures instructions and sent to a bio-analyzer to measure RNA concentration and quality. Reverse transcription was performed using a High-Capacity RNA-to-cDNA Kit (Life Technologies). cDNA was diluted and a master mix was prepared for Droplet Digital PCR (ddPCR).
ddPCR was run as a 20 µl reaction containing ddPCR Supermix for Probes (no dUTP) (Bio-Rad, Gladesville, NSW, Australia), cDNA or gDNA, 1 µl of each primer/probe (20×). The 20 µl sample, along with 70 µl of ddPCR-oil was loaded into a DG8 cartridge and covered with a gasket according to manufacturer’s instructions; the cartridge was then placed in a QX100 droplet generator. Into a 96 well PCR plate 40 µl of sample was transferred and then placed in a PCR thermocycler (Eppendorf, North Ryde, NSW, and Australia). The plate was then placed in the QX100 ddPCR reader (Bio-Rad, Gladesville, NSW, Australia) for quantification.
To measure C9ORF72, assays with FAM labels that detect each variant were used; mouse HPRT with a HEX label was used as an endogenous control (Table 2). Ratios were obtained by dividing absolute values from each variant to HPRT. Samples were normalized in respect to GFP controls. GraphPad Prism Program was used to graph and run One-way ANOVAs for three biological replicates.
To detect the mature amiRC9 product of rAAV9-H1-amiRC9-Cb-GFP, the RNA extracted from cells or tissue was further diluted to 10 ng/µl. cDNA was generated using the TaqMan microRNA reverse transcription kit (4366597; Life Technologies). We used a custom TaqMan small RNA assay that detects the mature artificial microRNA C9 (Table 2) and used small nuclear RNA135 as an endogenous control. For comparing the expression levels of amiRC9 with an endogenous microRNA, we used miR128a (Table 2), which is highly expressed in the brain.
Copy number variance (CNV) of the C9ORF72 transgene in BAC112 mice was measured as follows: brains were fresh frozen and cut in a sagittal section to extract DNA. Tissues were lysed overnight at 55 °C, RNAse treated and eluted following manufacturer’s instructions (Gentra Puregene Tissue Kit, Qiagen). The DNA was diluted to 20 ng/µl and 2 µl was added to the ddPCR master mix along with probes for genomic C9ORF72 and mouse EIF2C1 or RPP30 diploid genome. CNV was determined by dividing the absolute value of C9ORF72 by the absolute value of mEIF2C1 divided by 2 for diploid genome (Table 2). GraphPad Prism Program was used to graph the values. Homozygous mice have a CNV of 38–40, and heterozygous mice 18–20.
Western blot
Samples were lysed in RIPA buffer and cOmplete protease inhibitor (Roche), at 4 °C and the supernatant was collected. Five micrograms of protein lysates were run on Novex 12% Tris-Glycine gels (Life Technologies, USA) using Tris-Glycine SDS running buffer (Invitrogen, USA). Protein was transferred to nitrocellulose membranes using an i-Blot transfer device (Invitrogen, USA). Membranes were blocked for 1 h at room temperature with Odyssey Blocking Buffer (LiCor, USA) before being probed overnight with primary antibodies (Sigma ABN 1644–1645). IR labeled secondary antibodies were applied, and blots visualized. The band intensity was quantified using the Odyssey Infrared imaging system (LiCor, USA).
MSD ELISA
Samples were lysed in RIPA buffer and a cOmplete protease inhibitor tablet (Roche), using lysing Matrix D Beads (MP Bio) and a Tissue Lyser II (Qiagen 85300, 3 × 3 min 30HZS). Then, samples were placed in a new Eppendorf 1.5 ml tube and sonicated. After sonication, samples were centrifuged at 16,000 × g for 20 min and supernatants collected. The protein concentration of lysates was determined by a BCA assay (Thermo Scientific, USA). Poly-GP levels in lysates were measured using a previously described sandwich immunoassay that utilizes Meso Scale Discovery (MSD) electrochemiluminescence detection technology [28]. Lysates were diluted in Tris-buffered saline (TBS) and tested using 35 µg of protein per well in duplicates. Serial dilutions of recombinant (GP)8 in TBS were used to prepare the standard curve. Response values corresponding to the intensity of emitted light upon electrochemical stimulation of the assay plate using the MSD QUICKPLEX SQ120 were acquired, and background corrected using the average response from lysates obtained from non-transgenic mice prior to interpolation of poly-GP levels using a standard curve.
Fluorescent in situ hybridization
Twenty micrometers cryosections were serially collected (1/20) throughout the motor cortex and cervical spinal cord. Sections were fixed in 4% PFA for 20 min, followed by five 20-min washes in DEPC-treated PBS containing 0.1% Tween and 50 μg/ml of Heparin. Sections were pre-hybridized for 1 h at 55 °C in 2× sodium citrate (SSC) containing 0.1% Tween 20, 50% dextran sulfate, water, 50 μg/ml heparin, 1 mg/ml heat-denatured salmon sperm and 40% deionized sulfate. For hybridization, samples were incubated overnight at 55 °C in pre-hybridization buffer containing a 1:500 dilution of 1 μM DNA probe (sense: GGGGCC4) with a Cy3 label. After hybridization, the samples were washed in a pre-warmed mix 1:1 of hybridization buffer and in 2× SSC for 30 mins at 55 °C. This was followed by 2 washes in pre-warmed 2× SSC for 30 min at 55 °C, 2 washes with 0.2× SSC for 30 min at 55 °C, and 2 washes in PBS + 0.1%Tween + 50 μg/ml Heparin for 30 min, at room temperature. Sections were stained with DAPI (1:10,000) for 5 min, and auto fluorescence was quenched by incubation in 0.5% Sudan Black in 70% ethanol for 5 min. Glass coverslips were mounted using Lab Vision™ PermaFluor™ Aqueous Mounting Medium (Thermo Scientific Shandon, USA).
RNAscope fluorescent multiplex assay
Cervical cords were embedded in OCT, frozen and cryo-sectioned into 7-µm-thick sections and collected on plus/plus slides for staining. Sections were fixed in 10% NBF for 1 h, followed by two washes in PBS and four 5-min washes in ethanol (50%, 70%, 100%, and 100%) at room temperature. The fixed sections were covered with 5 drops of Protease IV for 30 min at room temperature followed by two washes in PBS prior to proceeding to the RNAscope Fluorescent Multiplex Assay protocol. Intron (RNAscope® Probe - C9orf72-intron1*E #405181) and exon (RNAscope® Probe - Hs-C9orf72 #405191) probes were warmed for 10 min at 400C, cooled to room temperature, and a 1:50 C2:C1 and C3:C1 hybridization mixture was prepared. Approximately 180 µl of hybridizing probe mixture was used to cover the sections completely, which was incubated for 2 h at 40 °C. Two 2 min washes in Wash Buffer (RNAscope Wash Buffer Reagents) with minor agitation preceded all of the Fluorescent Multiplex Detection Reagent additions. Approximately 180 µl of each of the fluorescent detection reagents was added to completely cover all sections within the hydrophobic barrier prior to incubation in the HybEZ™ Oven at 40 °C. A 30 min incubation was used for Amp 1-Fl, 15 min for Amp 2-Fl, 30 min for Amp 3-Fl, and 15 min for Amp 4-Fl-Alt-C. Sections were counterstained with DAPI for 30 s at room temperature and glass coverslips were mounted using Prolong Gold Antifade Mountant.
All images were taken using a Leica DM5500 B microscope with a Leica DFC365 FX camera. A total magnification of 100.8× (63× oil lens * 1.6× camera) was used to generate the images, while using the acquisition settings (Table 3) for the various probes. A maximum projection of z-stacks with a range of 1–3 µm was used for quantification. Approximately 15 images were taken from three different cervical spinal cord sections for each animal, each with at least one motor neuron present, n = 3 per group. The quantification of intron, exon, and colocalized targets was conducted manually for motor neurons in the ventral horn by RNAScope. Cervical spinal motor neurons were identified by location, morphology, and larger nuclei. Because tissues were fixed prior to staining, GFP expression was weak, rendering it impossible to count the GFP-positive cells (i.e. virally transduced cells) selectively. We therefore counted all RNA targets identified regardless of GFP transduction. Intron RNA targets were additionally classified as inside or outside of the nucleus by the presence of puncta within the DAPI signal. Co-localization of intron and exon puncta was determined using the z-stack and spectral color change as observed by light color properties of the RGB additive color model.
eGFP immunofluorescence
Mice were sacrificed by perfusion with PBS and 4% paraformaldehyde (PFA). The whole body was then immersed in 4% PFA. After overnight fixation, brains were dissected and cryo-preserved by being immersed in 10%, 20%, and 30% sucrose, overnight for each gradient, and embedded in OCT (Tissue-Tek; cat no. 4583). Tissues were cut into 40-μm coronal sections using a cryostat. Sections were stained with DAPI and antibodies against eGFP (Thermo Fisher Scientific; A-11122; 1:1000 diluted), NeuN (Millipore; MAB377; 1:1000 diluted), GFAP, (Abcam; ab4674; 1:1000) and Iba1 (Wako Chemicals; NC9288364; 1:1000) and proper fluorophore-labeled secondary antibodies (Invitrogen). Stained sections were mounted using Lab Vision™ PermaFluor™ Aqueous Mounting Medium (Thermo Scientific Shandon, USA). Immunofluorescence signal from sections was captured using an upright fluorescence microscope (Leica Microsystems).
Results
Design of amiRNA to target C9ORF72 variants
As a first step toward ameliorating the molecular hallmarks of the C9ORF72 HRE, we designed four amiRNAs targeting all variants of C9ORF72. The target location for the amiRNAs is depicted in Fig. 1B and Table 1. Efficacy of the amiRNAs to target C9ORF72 was screened in HEK293T cells, and mRNA levels for each variant were evaluated by quantitative Droplet Digital PCR (ddPCR). Expression of these four plasmids was driven by the chicken β-actin (CBA, polymerase II) promoter. The results were normalized to the GFP control. amiRC9 most consistently suppressed expression of all three C9ORF72 isoforms (Fig. 1C). Therefore, amiRC9 was cloned into a vector with the more powerful H1 promoter (polymerase III) [29] and packaged into rAAV9 vector and used in all subsequent in vitro and in vivo experiments.

A Schematic of the artificial microRNA plasmid constructs: The first plasmid was used for preliminary screening in HEK293T and the second for further experiments in vivo. 1. Chicken beta actin promoter driving enhanced Green Fluorescent Protein (eGFP) and the artificial microRNA, followed by a PolyA tail and flanked by inverted terminal repeats (ITRs). 2. H1 promoter driving the expression of the artificial microRNA followed by a CBA promoter driving eGFP a PolyA tail. Flanking ITRs are noted. B Target location of each artificial microRNA; each amiR is labeled by its nucleotide target. Transcript-specific alternative first exons are shaded. C Screening of microRNAs in human HEK293T cell line: HEK293T cells were transfected with 4 artificial microRNA plasmids driven by the CBA promoter. Plots show ddPCR analyses of expression of each individual variant V1, V2, V3 as well an assay detecting all variants jointly (Vall), normalized to human HPRT and compared to the GFP control. Mean ± SEM, n = 3, biological replicates, One-way ANOVA, Bonferroni: p* 0.01–0.05; p** 0.001–0.01. D Primary culture timeline: Male homozygous mice were crossed with wild type female C57/BL6 mice. Cortical neurons were dissected from multiple embryos at embryonic day 15, pooled and plated at 50,000 cells/cm2 and infected with each vector at day in vitro 4 (DIV4) at a MOI of 50,000 AAV. Three to five technical replicates were performed per condition and collected at (DIV10). E Digital PCR analysis of expression of each individual variant V1, V2, V3 as well an assay detecting all variants jointly (Vall), normalized to mouse HPRT and compared to the GFP control. amiRC9 decreases each variant by ~50% in 2 independent experiments with 3-4 technical replicates each. Mean ± SEM, n = 2, biological replicates, One-way ANOVA, Bonferroni: p* 0.01–0.05; p** 0.001–0.01; p*** 0.0001–0.001; p****<0.0001. F Comparison of levels of mature amiRC9 normalized to small nuclear RNA13 (mean ± SEM). miR128a, which is highly expressed in the brain. G Concentration of poly-GP measured by ELISA in mixed cortical neurons from the same 2 biological experiments in (E) consistent decrease in treated samples. Mean ± SEM, n = 2, biological experiments, One-way ANOVA, Bonferroni: p** 0.001–0.01; p*** 0.0001–0.001. H Fluorescent in situ hybridization quantification of the sense RNA foci in 4-well chamber slides from the second set of primary cultures (Exp2). The graph quantifies foci in 2–3 chambers with ~300 cells counted. Grouped into cells with 1–10 foci, 11–40 or greater than 40. Two-way ANOVA, no significant differences between treated and untreated groups.
amiRC9 silences C9ORF72 in vitro
We next assessed silencing by AAV9-H1-amiRC9 in primary cortical cultures from the BAC112 mouse model [27]. This mouse model harbors the full-length human C9ORF72 gene with an expansion of 550 repeats. Heterozygous mice have 16–20 copies of the transgene and homozygous 32–40 copies. This model recapitulates the pathological molecular aspects of the disease, including RNA foci and DPRs (Supplementary Fig. 1). Two independent cultures were transduced with a GFP control vector, a PBS-mock control, and the amiRC9 vector at day in vitro (DIV) 4 at a multiplicity of infection (MOI) of 50,000 vector particles. The cultures were harvested at DIV10 (Fig. 1D), and total RNA was extracted. ddPCR quantification revealed that amiRC9 significantly reduced each variant of the C9ORF72 mRNA (Fig. 1E).
To further confirm that C9ORF72 silencing reflected post-transcriptional regulation of gene expression by amiRC9, we designed a custom miRNA assay to quantify the mature form of amiRC9 levels by ddPCR (Fig. 1F). As expected, only treated samples had detectable levels of amiRC9. In addition to detecting amiRC9, we wanted to compare the expression levels to other endogenous miRNAs. We therefore assayed levels of microRNA128a, which is highly expressed in brain [30, 31] and found that amiRC9 is 19–25-fold less abundant than miR128a. This provides assurance that the observed level of amiRC9 is not likely to be saturating the RNAi machinery or inducing toxicity by competing with endogenous miRNAs entering the RISC complex.
We had previously shown that amiRC9 can also decrease poly-GP DPRs in primary neuronal cultures [32]. To determine if we could attain a similar reduction in cultured neurons derived from a high copy transgenic mouse model, we quantified the poly-GP levels in treated and control neuronal cultures derived from the BAC112. Notably, the MSD ELISA assay showed the GP DPRs were significantly reduced in treated cultures (Fig. 1G), in alignment with our previous results [32].
While microRNAs primarily act in the cytoplasm [33,34,35], published data [36,37,38] suggest that they can be shuttled back into the nucleus and thereby decrease the number of RNA foci [39]. To test this possibility, we used fluorescent in situ hybridization (FISH) to detect sense foci. Multiple 4-well chamber slides were scored for foci number per nucleus (Fig. 1H). Quantification revealed no significant differences in numbers of intranuclear foci between treated and control samples, suggesting that amiRC9 had not suppressed C9ORF72 transcripts inside the nucleus.
Striatal brain injections silence C9ORF72 variants in vivo
Multiple lines of transgenic BAC mouse models have been generated [21, 27, 32, 40,41,42] that differ with respect to mouse background, expansion size, transgene insertion site, copy number, and C9ORF72 expression levels. While these models recapitulate the molecular hallmarks C9ORF72 in humans (RNA foci and toxic DPRs of C9ORF72-linked ALS), they do not recapitulate motor neuron loss and paralysis. For in vivo studies, we elected to use the BAC112 transgenic mouse model created by the Baloh Lab [27].
To determine its efficacy in vivo, we first delivered AAV9-H1-amiRC9 vector into adult heterozygous mice via tail vein injection. Two weeks post injection, liver tissues were collected C9ORF72 mRNA levels were analyzed by ddPCR (Supplementary Fig. 2). We found a 50% mRNA decrease of all three C9ORF72 variants in the liver. After confirming the vector’s effectiveness, we next injected the vector directly into the striatum of adult homozygous BAC112 mice. We opted to use striatal injections because they are a comparatively easy method for injecting a large volume of vector to a well-defined and relatively circumscribed region of the brain.
Homozygous 4-month-old mice were gender and copy number matched and separated into three injection groups: PBS-mock, AAV9-CB-GFP control, and AAV9-H1-amiRC9. We performed bilateral striatal injections, delivering 5 µl of vector per side for a final dose of 1E11 genomic copies (GC) per mouse (Fig. 2A). Mice were culled 16 weeks post injection, PBS-perfused, and dissected. Two-millimeter punches were taken from each hemisphere for RNA and protein extraction. As in treated primary culture experiments, we found significant diminution of each of the C9ORF72 mRNA variants (~50% reduction) when comparing amiRC9 to GFP control and PBS-mock injected mice (Fig. 2B). This was consistent with the presence of significant levels of the mature form of amiRC9 in treated samples only (Fig. 2C).

A Striatal brain image from an injected mouse depicting the injection coordinates. Injections were performed 2 mm from the midline and 1 mm from bregma at a depth of 2 mm. The arrow depicts ~2 mm punch biopsy location. B Starting from a 2 mm punch dissected from the striatum we ran ddPCR analysis of expression of each individual variant (V1, V2, V3) as well an assay detecting all variants jointly (Vall), normalized to mouse HPRT and referenced to the GFP control. Approximately ~50% silencing is observed in H1-amiRC9 treated mice. Mean ± SEM, mice n = 4 CB-GFP, n = 7 PBS, n = 7 H1-amiRC9, One-way ANOVA, Bonferroni: p** 0.001–0.01; p*** 0.0001–0.001; p****<0.0001. C Comparison of levels of mature amiRC9 and miR128a, normalized to the small nuclear RNA135 (mean ± SEM). D Western immunoblot image for the long isoform (55 kDa) protein of C9ORF72, normalized to actin ~42 kDa. Shown from left to right are KO mouse sample used as a negative control (lane 1), C57 WT mouse controls (lanes 2–4), PBS (lanes 5–7) and GFP (lanes 8–10) controls and H1-amiRC9 treated mice (lanes11–14). There is a clear decrease of signal in mice treated with H1-amiRC9. E Densitometric quantification of western blot in (D); Levels from wild-type mice (n = 3) were averaged and subtracted as a background signal. Significant silencing is detected compared to untreated controls. One-way ANOVA, Bonferroni: p** < 0.001-–0.01. F ELISA-based protein levels of poly-GP from groups corresponding to groups in (E). H1-amiRC9 treatment reduced levels ~50%. Mean ± SEM, One-way ANOVA, Bonferroni: p** 0.001–0.01; p****<0.0001.
To quantify the protein levels of C9ORF72, we performed Western blot studies using an antibody with relative specificity for human C9ORF72. We were able to detect the mouse C9ORF72 ortholog, which was not detected in brain samples from C9ORF72 knockout mice [43]. To control for endogenous mouse C9orf72 detection, we ran three wild type C57BL/6 brain samples side-by-side the treated samples and controls. With densitometry, we quantified protein levels and subtracted the average of the WT mouse samples as background. Protein levels of the long isoform (481 aa ~51 kDa) (Fig. 2D, E) was significantly decreased by more than 50% in treated mice.
Having observed efficient knockdown of C9ORF72 transcript and protein levels, we next sought to determine whether this RNAi-mediated knockdown would reduce toxic DPRs, one of the major pathogenic gain-of-function hallmarks of C9-ALS/FTD. Using an immunoassay that detects poly-GP, we detected a 50–60% reduction of GP DPRs in treated mice compared to controls (Fig. 2F).
CSF-based and peripheral delivery yield comparable transduction of the rostral CNS and spinal cord
Multiple delivery routes provide therapeutic access to the neuroaxis. To optimize suppression of C9ORF72 expression through amiRC9, we compared the intracerebroventricular (ICV) injection route to the peripheral intravenous (IV) temporal (facial) vein route in neonatal mice.
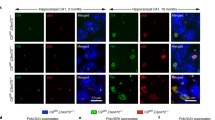
Heterozygous P1 pups were bilaterally ICV injected [44] with a total of 2E10 GC of AAV9-H1-amiRC9 or AAV9-CB-GFP, or an equal volume of PBS (Fig. 3A). At 16 weeks post injection, the whole brain and spinal cords were harvested from treated and control mice. Frontal lobes were dissected and freshly frozen for RNA extraction or immunofluorescence staining. We assessed knockdown for each variant as described above. There was silencing of all C9ORF72 isoforms in the treated samples (Fig. 3B), which reached statistical significance for V2, V3, and total C9ORF72 transcripts. We documented the presence of the mature amiRC9 in all injected animals and quantitated levels via ddPCR (Fig. 3C). By performing immunofluorescent co-staining in the brain, we confirmed that transduction of AAV9-H1-amiRC9-GFP occurred in neurons and astrocytes, but we noted a lack of co-localization of GFP with microglia, as defined by immunostaining with the antibody Iba1 (Supplementary Fig. 3). These results are consistent with other reports using AAV9 to transduce the CNS [45]. By ELISA, we also observed a trend toward a decrease in GP DPRs in these frontal lobe preparations (Fig. 3D). Using FISH analysis, we did not detect significant differences in the number of sense RNA foci in layer V of the frontal cortex (Fig. 3E, F).

A Cranium of neonatal BAC112 transgenic mice illustrating the ICV Injection sites (black arrows). As references, the eye positions are indicated by asterisks. B Neonatal mice were injected bilaterally with 2 µl of vector at ~1E13GC and sacrificed at 16 weeks post injection. Total RNA from frontal lobe tissue was used for Digital PCR analysis of expression of each individual variant (V1, V2, V3) and all variants jointly (Vall). For each, transcript levels were normalized to mouse HPRT and referenced to the GFP control. Mice treated with the H1 promoter showed significant suppression. Mean ± SEM, mice n = 8 CB-GFP, n = 6 PBS, n = 4 H1-amiRC9; One-way ANOVA, Bonferroni: p** 0.001–0.01; p*** 0.0001–0.001; p****<0.0001. C Detection of mature amiRC9 product, normalized to small nuclear RNA135 (mean ± SEM) and compared to endogenous microRNA128a (also normalized to sno125). D ELISA-based protein levels of poly-GP showed a trend towards silencing by amiRC9 when compared to mock-treated PBS controls and untreated controls (for which there was greater scatter of data). E Fluorescent in situ hybridization confirms the presence of sense RNA foci in layer V of the frontal lobe of treated and control mice. RNA foci were not detected following RNAse treatment or in non-transgenic controls. F Quantification of RNA foci in layer V of the frontal lobe showed no significant differences between treated and untreated groups. The graph quantifies an average of 3 mice per group, ~500 cells counted per 3 images. Grouped into cells with 1–5 foci, 6–10 or greater than 10. Two-way ANOVA, no significant differences between treated and untreated groups.
We next tested the efficacy of temporal vein injections. P1 pups were injected with 50 µl of vector at a dose of 2E11 GC (Fig. 4A). At 16 weeks post injection, we investigated C9ORF72 mRNA expression in the frontal lobes and found detectable levels of the mature amiRC9a but no significant decrease of C9ORF72 mRNA (Supplementary Fig. 4), which is in contrast with the C9ORF72 mRNA silencing we observed in the frontal lobe of ICV-injected pups (Fig. 3B).

A Temporal vein injection site depicted in neonatal mice. Pups were injected 50 µl of each vector at 2E11vp/ml into the temporal vein. Mice were sacrificed at 16 weeks post injection. B After temporal vein injection, total RNA from laser-captured motor (LCM) neurons from the cervical portion of the cord showed significant silencing of Vall. Samples were normalized to mouse HPRT and referenced to the GFP control. Mean ± SEM, mice n = 4 CB-GFP, n = 4 PBS, n = 6 H1-amiRC9; one-way ANOVA, Bonferroni: p*** 0.0001–0.001; p**** < 0.0001. C Comparison in laser-captured motor neurons of levels of mature amiRC9 and microRNA128 (both normalized to small nuclear RNA125) following temporal vein injections (mean ± SEM). D ICV-injected mice were described in Fig. 3. After ICV injection, total RNA from laser-captured motor (LCM) neurons from the cervical portion of the cord showed significant silencing of Vall. Samples were normalized to mouse HPRT and referenced to the GFP control. Mean ± SEM, mice n = 4 CB-GFP, n = 4 PBS, n = 6 H1-amiRC9; one-way ANOVA, Bonferroni: p* 0.01–0.05; p** 0.001–0.01. E Comparison in laser-captured motor neurons of levels of mature amiRC9 and microRNA128 (both normalized to small nuclear RNA125) following ICV injections (mean ± SEM).
While the efficacy of these delivery methods was distinguishable for the frontal cortex, we anticipated that systemic delivery would provide greater mRNA silencing in the spinal cord [44]. To test this, we next investigated which of these two delivery methods most efficiently silenced C9ORF72 in the spinal cord. We assayed C9ORF72 expression in motor neurons specifically using laser capture microdissection (LCM) of motor neurons within the ventral horn of cervical cord sections. We then quantified both the mRNA levels of the C9ORF72 variants and the presence of the mature amiRC9. Temporal vein delivery achieved only slightly better suppression of C9ORF72 (~50%, Fig. 4B, C), than ICV delivery (30–35% decrease, Fig. 4D, E).
amiRC9 targets misspliced transcript that produces the cytoplasmic toxic DPRs
We next sought to define the mechanism by which amiRC9 decreases levels of toxic DPRs. We anticipated that, in general, there should be both intronic and exonic targets for silencing of RNA in the nucleus. We further anticipated that only the exonic targets would be present in the cytoplasm. In the case of mutant C9ORF72, however, the existence of the DPRs suggests that there is a fragment of misspliced intronic RNA that is present not only in the nucleus, but also in the cytoplasmic compartment. Indeed, it has further been reported that intron-HRE fragments are present in the cytoplasm of C9ORF72 motor neurons [46, 47]. It is also possible that such intron-HRE fragments might contain exons as well. If the intron-HRE and any exons are retained within the misspliced transcript, we expected to see these colocalize in both the nucleus and the cytoplasm. We further expected that amiRC9 would decrease levels of both intron and exon targets in the cytoplasm of treated cervical cord samples. To evaluate this hypothesis, we used RNA in situ hybridization (RNAscope) on cervical cord sections from the temporal vein-injected group and designed two probes: the first targeting the intronic area just before the HRE (intron target) and the second targeting spliced joint exons 3–6 (exon target) (Fig. 5A). Ventral horn motor neurons were imaged as Z-stacks and each punctum detected was quantified as an RNA target. Representative images in (Fig. 5B) illustrate results from the three different groups: mock, control, and H1-amiRC9 treated cervical spinal cords. The first column shows DAPI staining of the nucleus of the cell followed by the exon target in red, the intron target in green, and the overlay in yellow. Dotted areas on the top right corners emphasize magnified cytoplasmic regions containing colocalized targets. We scored for total exon targets, cytoplasmic intron targets, and colocalized targets (Fig. 5C). Treatment with AAV9-amiRC9 resulted in a ~40% decrease of total exon targets, ~60% cytoplasmic intron targets, and ~50% of colocalized targets. We also identified the presence of cytoplasmic intron targets that did not colocalize with the exon target. We hypothesize that these are the unaccompanied intron-HRE transcript, which cannot be targeted by amiRC9, and which contribute to the generation of the DPRs.

A Representation of target locations for RNAscope probes hybridizing to an intronic sequence just upstream of the HRE and an exon sequence spanning exons 3–6. B Cervical cord sections were cut at 10 µm and incubated with multiplex probes detecting C9ORF72 Intron1 (Cy3-Green) and C9ORF72 Exons2-6 (Cy5-red). The nuclei are stained with DAPI. Each row contains representative images from controls (GFP, PBS) and treated animals (AAV9-H1-amiRC9). Motor neurons were identified based on morphology and position within the ventral. The right-most panel column shows the co-localization of both color channels; merged signals are yellow. Quantification of each target RNA for independent channels shown in panel. The treated samples are distinguishable from controls by a significant reduction in the RNA target signal for each channel. C Quantification of RNA targets in motors neurons in the ventral horn of cervical cords. Each RNA target was manually quantified using z-stacks and the total exon target was tallied. Each target was normalized to the average total quantification of each control target. 3 biological replicates and 1 technical replicate were used per condition, encompassing a total of 5 pictures per animal. Mean ± SEM; one-way ANOVA, Bonferroni: p* 0.01–0.05; p** 0.001–0.01; p****<0.0001.
Discussion
Recent publications from our group and others have documented that AAV-mediated delivery of amiRNAs can suppress expression of mutant genes that cause disorders such as Huntington’s disease and ALS [32, 44, 48,49,50]. Here we designed and tested multiple amiRNAs targeting exonic regions within the C9ORF72 gene. After screening in HEK293T cells, we selected the top silencing candidate and further validated its efficiency in primary cortical cultures and in vivo in a C9ORF72 transgenic mouse model. This model overexpresses C9ORF72 from multiple tandem copies of the transgene [27]. However, in a clinical setting most patients carry a single copy of the C9ORF72-HRE mutation, and only a few cases are homozygous. Mindful of these differences, we set out to demonstrate that our therapeutic strategy could silence C9ORF72 and its toxic counterparts sustainably.
The primary neuronal culture experiments confirmed the effectiveness of rAAV9-amiRC9 at transducing cortical neurons shown by eGFP expression, proper processing of amiRC9 into mature miRNA, and reduction of C9ORF72 mRNA and DPR levels. As anticipated, numbers of intranuclear RNA foci were not reduced; like most miRNAs, amiRC9 appears not to shuttle back into the nucleus [36,37,38, 51].
We next tested the efficiency of amiRC9 in vivo, by performing stereotaxic striatal injections in adult mice to deliver a high dose of vector to a broad area of the brain. We confirmed proper distribution of the vector into the striatum by eGFP expression, where medium spiny neurons compose most of the neuronal population transduced. Significant silencing of HRE-retaining C9ORF72 mRNA resulted in a ~50% suppression of GP DPRs, supporting the view that AAV9-mediated amiRC9 delivery in adult mice successfully suppressed C9ORF72 and the toxic byproducts of the HRE.
We compared two different delivery approaches in neonatal mice: local delivery with ICV injections and systemic delivery through the temporal vein. ICV injection significantly reduced C9ORF72 variant mRNA isoforms and revealed a trend toward reduction of GP DPRs. By contrast, the temporal vein injection did not reduce the C9ORF72 mRNA (Supplementary Fig. 4). This may reflect a dilution effect of dissemination of the virus in the venous circulation. To investigate C9ORF72 suppression specifically in motor neurons, we used LCM in the cervical spinal cord. We found significant silencing in the motor neurons of both ICV and temporal vein-injected mice, but remarkably, most silencing was achieved in the motor neurons of temporal vein-injected mice.
Assuming that most cells are transduced, and the majority of silencing is post-translational, we hypothesized that some fraction of C9ORF72 mRNA is located in the nucleus and therefore is inaccessible to amiRC9. Similarly, any misspliced intron-HRE RNA or intron-HRE-exon RNA that is retained in the nucleus will also be inaccessible to amiRC9. By contrast, species of misspliced intron-HRE-exon 3 containing RNA in the cytoplasm would be targeted by amiRC9 and, moreover, should show co-localization of probes to the intron and the associated exons. Because amiRC9 targets exon 3, any exon-free intron-HRE RNA in the cytoplasm should not be suppressed by amiRC9. To address these questions of localization, we performed in situ hybridization using specific probes targeting either the first intron or the joint exons 3–6. We detected the exon RNA target in both cellular compartments at a nuclear to cytoplasmic ratio of ~2:3. Quantification of the exon target showed it to be significantly reduced only in the cytoplasm of treated mice.
Normally, one would expect intron-containing mRNA to be nuclear or perhaps perinuclear. Our observation that they are detected broadly in the cytoplasm is consistent with the concept that it is this misspliced intron-HRE RNA that participates in atypical translation of the HRE. The infrequent, colocalized exon-intron transcripts were significantly decreased in treated mice. This indicates that these abnormal transcripts containing exon 3 and HRE are accessible for amiRC9 targeting. On the other hand, the cytoplasmic intron-HRE RNAs that do not colocalize with the exon target cannot be targeted by amiRC9 and are likely the source of the residual poly-GP production that is not diminished by amiRC9 treatment. Other publications [52, 53] similarly document the presence of the retained intron-HRE and additionally demonstrate that there is a potential translational start site (CUG) upstream of the intron, driving the translation of the HRE [54]. Further aspects of the expansion, however, remain to be studied. We cannot exclude the possibility that a portion of the HREs remains within a lariat or as an unaccompanied intron-HRE transcript. In Drosophila and patient motor neurons, it has been shown that sequestration of SRSF1 onto repeat transcripts overcomes nuclear retention by driving the nuclear export of pre-mRNAs retaining exon-1 and pathological expansions in intron-1; depletion of SRSF1 blunts export and reduces levels of dipeptide repeat proteins [55].
These considerations and an overview of the mechanism of action of amiRC9 are summarized in Fig. 6. The first two steps depict the C9ORF72 gene and the 3 variant transcripts it produces. Variants 1 and 3, which are less abundant, harbor the HRE in the first intron. When missplicing occurs, variants 1 and 3 retain the intron and the HRE, which is exported to the cytoplasm, permitting repeat-associated translation of toxic DPRs [22]. With rAAV9-mediated delivery of amiRC9, the primary microRNA enters the RNAi silencing pathway, is exported to the cytoplasm to be further cleaved to enter the RISC complex and binds in perfect complementarity to the target mRNA. Since amiRC9 targets exon 3, both properly spliced C9ORF72 mRNA and intron-HRE-exon 3 retaining mRNA are targeted for silencing.

1 The C9ORF72 gene produces three different variants. Variants 1 and 3 include the hexanucleotide repeat expansion, depicted by the red diamond within the first intron. 2 With proper splicing, variant 1 produces a short 221 amino acid (~24 kDa) protein while variants 2 and 3 generate a long 481 amino acid (~55 kDa) protein. 3 When unconventional splicing occurs, variants 1 and 3 retain the intron along with the hexanucleotide expansion. The presence of the expansion in the cytoplasm permits repeat-associated non-ATG translation (RAN) generating three DPRs from the sense strand (GA, GR, GP) and three from the antisense strand (GP, PR, PA). 4 After rAAV9-mediated delivery of amiRC9, the primary microRNA enters the RNAi silencing pathway. It is first trimmed by the microprocessor complex Drosha, and then exported to the cytoplasm to be further cleaved by Dicer. The mature microRNA will enter the RISC complex and bind in perfect complementarity to the target mRNA for cleavage. Since amiRC9 specifically targets exon 3, both properly spliced and intron retaining C9ORF72 mRNA transcripts will be targeted for silencing. This results in suppression of ~ 50% of the C9ORF72 mRNA, protein and GP DPRs both in vitro and in vivo.
While the approach described here targeted all C9ORF72 transcript variants, another would be to design amiRNAs that target either the expansion carrying V1 and V3, or the intron-HRE for both sense and antisense strands. Because V1 and V3 represent a small fraction of total C9ORF72 mRNA, targeting V1 and V3 should spare the abundant normal transcript V2 and thus preserve levels of the C9ORF72 protein. Recently, encouraging data using different amiR scaffolds and AAV5 as a delivery vehicle also support the use of AAV-mediated amiR silencing as a safe and reliable approach for the treatment of ALS [39, 56]. Antisense oligonucleotides (ASOs) targeting V1 and V3 are an alternative strategy to target C9ORF72, with encouraging early results in small clinical trials [57, 58]. An advantage of ASOs is their capability to penetrate the nucleus, although a drawback is the requirement for recurrent treatments, which in combination with the challenges of direct delivery in the CNS, adds additional strain on already physically vulnerable ALS patients [59]. AAV has the advantage of long-term expression of cargo in post-mitotic cells such as neurons [60]. This renders AAV-mediated delivery of amiRNAs a particularly appropriate strategy for the treatment of ALS. In previous studies we explored the use of AAV-mediated amiRNAs targeting SOD1 and found significant reductions of SOD1 and preservation of motor neurons in treated animals and in a human study treating two SOD1-ALS patients [50].
References
Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–30.
Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
DeJesus-Hernandez M, Mackenzie IRR, Boeve BFF, Boxer ALL, Baker M, Rutherford NJJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. 2011;72:245–56.
Majounie E, Renton AE, Mok K, Dopper EGPP, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–30.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68.
Devenney E, Hornberger M, Irish M, Mioshi E, Burrell J, Tan R, et al. Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol. 2014;71:331–9.
Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev. 2017;44:117–24.
Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathologica. Communications. 2019;7:115.
Amick J, Ferguson SM. C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 2017;18:267–76.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24:313–25.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23:615–24.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845–57.
Gendron TF, Bieniek KF, Zhang YJJ, Jansen-West K, Ash PEAA, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–44.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–8.
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–77.
Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin WL, DeJesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–25.
Sun Y, Eshov A, Zhou J, Isiktas AU, Guo JU. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat Commun. 2020;11:3354.
West RJH, Sharpe JL, Voelzmann A, Munro AL, Hahn I, Baines RA, et al. Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in Drosophila. Acta Neuropathol Commun. 2020;8:158.
Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363:eaav2606.
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24:1136–42.
Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;2012:CD001447.
Takei K, Takahashi F, Liu S, Tsuda K, Palumbo J. Post-hoc analysis of randomised, placebo-controlled, double-blind study (MCI186-19) of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:49–54.
Cruz MP. Edaravone (Radicava): a novel neuroprotective agent for the treatment of amyotrophic lateral sclerosis. P T. 2018;43:25–8.
Aschenbrenner DS. New drug approved for ALS. Am J Nurs. 2023;123:22.
O’Rourke JG, Bogdanik L, Muhammad AKMG, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88:892–901.
Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron. 2017;94:1056–70.
Borel F, Gernoux G, Sun H, Stock R, Blackwood M, Brown RH, et al. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci Transl Med. 2018;10:eaau6414.
Bak M, Silahtaroglu A, Møller M, Christensen M, Rath MF, Skryabin B, et al. MicroRNA expression in the adult mouse central nervous system. RNA. 2008;14:432–44.
Adlakha YK, Saini N. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol Cancer. 2014;13:33.
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88:902–9.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98.
Ambros V. The evolution of our thinking about microRNAs. Nat Med. 2008;14:1036–40.
Hoffer P, Ivashuta S, Pontes O, Vitins A, Pikaard C, Mroczka A, et al. Posttranscriptional gene silencing in nuclei. Proc Natl Acad Sci USA. 2011;108:409–14.
Huang V, Li LC. miRNA goes nuclear. RNA Biol. 2012;9:269–73.
Nishi K, Nishi A, Nagasawa T, Ui-Tei K. Human TNRC6A is an Argonaute-navigator protein for microRNA-mediated gene silencing in the nucleus. RNA. 2013;19:17–35.
Martier R, Liefhebber JM, Miniarikova J, van der Zon T, Snapper J, Kolder I, et al. Artificial microRNAs targeting C9orf72 can reduce accumulation of intra-nuclear transcripts in ALS and FTD patients. Mol Ther Nucleic Acids. 2019;14:593–608.
Batra R, Lee CW. Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/ frontotemporal dementia. Front Cell Neurosci. 2017;11:196.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–50.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90:521–34.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra93.
McLean JR, Smith GA, Rocha EM, Hayes MA, Beagan JA, Hallett PJ, et al. Widespread neuron-specific transgene expression in brain and spinal cord following synapsin promoter-driven AAV9 neonatal intracerebroventricular injection. Neurosci Lett. 2014;576:73–8.
Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS ONE. 2013;8:e76310.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65.
Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabreja GC, Song L, et al. Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1G93A mice and nonhuman primates. Human Gene Therapy. 2016;27:19–31.
Pfister EL, Chase KO, Sun H, Kennington LA, Conroy F, Johnson E, et al. Safe and efficient silencing with a Pol II, but not a Pol lII, promoter expressing an artificial miRNA targeting human huntingtin. Mol Ther Nucleic Acids. 2017;7:324–34.
Pfister EL, DiNardo N, Mondo E, Borel F, Conroy F, Fraser C, et al. Artificial miRNAs reduce human mutant huntingtin throughout the striatum in a transgenic sheep model of Huntington’s disease. Hum Gene Ther. 2018;29:663–73.
Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med. 2020;383:151–8.
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877.
Sznajder ŁJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc Natl Acad Sci USA. 2018;115:4234–9.
Niblock M, Smith BN, Lee YB, Sardone V, Topp S, Troakes C, et al. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta Neuropathol Commun. 2016;4:18.
Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZZ, et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun. 2018;9:152–152.
Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez-Martinez A, Cooper-Knock J, Higginbottom A, et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun. 2017;8:16063.
Martier R, Liefhebber JM, García-Osta A, Miniarikova J, Cuadrado-Tejedor M, Espelosin M, et al. Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther Nucleic Acids. 2019;16:26–37.
Liu Y, Andreucci A, Iwamoto N, Yin Y, Yang H, Liu F, et al. WVE-004, an investigational stereopure antisense oligonucleotide for the treatment of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (2302). Neurology. 2021;96. https://n.neurology.org/content/96/15_Supplement/2302
Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28:117–24.
Meijboom KE, Brown RH. Approaches to gene modulation therapy for ALS. Neurotherapeutics. 2022;19:1159–79.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Acknowledgements
We acknowledge G. Gao and Q. Su at University of Massachusetts Medical School who supervised the vector production. We thank Lorelei Stoica for training on all mouse surgeries, Lina Song for aiding with plasmid cloning and instruction, Qiushi Tang for assisting with vector constructs, Meghan Blackwood for aiding with tissue collections, Ozgun Uyan for scientific conversations and suggestions, and Allison Keeler for scientific conversations and editing the manuscript.
Funding
This study was supported by ALS Alliance Therapy (RHB), the ALS-FindingACure (RHB), and NINDS NS088689 (C.M. and RHB). RHB also received funding from the NINDS (NS079836), the ALS Association, ALSOne, the Angel Fund, the Cellucci Fund for ALS Research, the Michael Rosenfeld ALS Fund, the Pierre L. de Bourgknecht ALS Research Foundation, Project ALS and Target ALS.
Author information
Authors and Affiliations
Contributions
GTC led design, analysis, interpretation of the study, and prepared the manuscript. Performed screenings, cultures, surgeries, ddPCR analysis, RNA FlSH and quantifications. KEM prepared manuscript for publication, did analyses and statistics and performed immunohistochemistry and microscopy experiments. AA aided with surgeries and temporal injections, primary cultures, scientific input, and editing. HT performed MSD ELISA assays for DPR detection, aided with scientific input. ZF performed animal tissue collection and sectioning including RNAscope quantification. AW managed the mice colony and aided with stereotaxic surgeries. NW performed Western blots for detection of C9ORF72 protein. RS performed cryosections of spinal cord and LCM microdissections. TG performed ELISA assays for DPR detection. AGr aided with temporal injections and manuscript editing. AGi aided with primary culture dissection. RHB and CM conceived of the project and supervised all aspects of its execution and analysis.
Corresponding authors
Ethics declarations
Competing interests
RHB and CM are inventors on the patent for the technology described within this report (rAAV-Based Compositions and Methods for Treating Amyotrophic Lateral Sclerosis, WO2015143078A1) and may be entitled to royalty payments in the future. The patent has been licensed to Apic-Bio for which CM and RHB are founders with equity. All other authors declare no competing financial interests.
Ethical approvals
All mouse experiments were conducted at UMass Medical School following protocols approved by the Institutional Review Board. The University of Massachusetts Medical School Institutional Animal Care and Use Committee approved all experiments involving animals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cabrera, G.T., Meijboom, K.E., Abdallah, A. et al. Artificial microRNA suppresses C9ORF72 variants and decreases toxic dipeptide repeat proteins in vivo. Gene Ther 31, 105–118 (2024). https://doi.org/10.1038/s41434-023-00418-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-023-00418-w
