
Abstract
Biallelic loss-of-function variants in TBC1D2B have been reported in five subjects with cognitive impairment and seizures with or without gingival overgrowth. TBC1D2B belongs to the family of Tre2-Bub2-Cdc16 (TBC)-domain containing RAB-specific GTPase activating proteins (TBC/RABGAPs). Here, we report five new subjects with biallelic TBC1D2B variants, including two siblings, and delineate the molecular and clinical features in the ten subjects known to date. One of the newly reported subjects was compound heterozygous for the TBC1D2B variants c.2584C>T; p.(Arg862Cys) and c.2758C>T; p.(Arg920*). In subject-derived fibroblasts, TBC1D2B mRNA level was similar to control cells, while the TBC1D2B protein amount was reduced by about half. In one of two siblings with a novel c.360+1G>T splice site variant, TBC1D2B transcript analysis revealed aberrantly spliced mRNAs and a drastically reduced TBC1D2B mRNA level in leukocytes. The molecular spectrum included 12 different TBC1D2B variants: seven nonsense, three frameshifts, one splice site, and one missense variant. Out of ten subjects, three had fibrous dysplasia of the mandible, two of which were diagnosed as cherubism. Most subjects developed gingival overgrowth. Half of the subjects had developmental delay. Seizures occurred in 80% of the subjects. Six subjects showed a progressive disease with mental deterioration. Brain imaging revealed cerebral and/or cerebellar atrophy with or without lateral ventricle dilatation. The TBC1D2B disorder is a progressive neurological disease with gingival overgrowth and abnormal mandible morphology. As TBC1D2B has been shown to positively regulate autophagy, defects in autophagy and the endolysosomal system could be associated with neuronal dysfunction and the neurodegenerative disease in the affected individuals.
Similar content being viewed by others

Introduction
Biallelic loss-of-function variants in TBC1D2B have previously been linked with a complex and evolving phenotype in three unrelated patients [1]. In the initial three cases, a characteristic phenotypic pattern was evident, encompassing neurodevelopmental and neurodegenerative traits, along with distinctive features of facial and gingival overgrowth. Specifically, a boy and a girl presented with mild developmental delay and mild intellectual disability, respectively. The third subject developed normally until 12–13 years of age and then showed mental deterioration that was accompanied by slurred speech and gait ataxia. In late adolescence, he developed visual deterioration, flexion contractures, and limb tremor. Seizures occurred in all subjects. Gingival overgrowth became apparent in early childhood. Cherubism was identified in the girl through the presence of enlargement of upper and lower jaws and multilocular, radiolucent lesions of the mandible in the orthopantogram [1]. Subsequently, two adult brothers carrying a homozygous TBC1D2B 1-bp deletion were reported with a similar clinical course [2].
TBC1D2B encodes a member of the family of Tre2-Bub2-Cdc16 (TBC)-domain containing RAB-specific GTPase activating proteins (TBC/RABGAPs) that are key regulators of RAB proteins [3, 4]. Members of the family of RAB small GTPases switch between an active, GTP-bound and an inactive, GDP-bound state and are required for endolysosomal membrane trafficking [5]. TBC1D2B binds various RAB proteins, including RAB31, and can transform active RAB proteins into their inactive forms through its GTPase activating domain [6,7,8]. In HeLa cells, TBC1D2B was found to co-localize with vesicles positive for RAB5 and partially with EEA1, both markers of early endosomes [1]. To date, three nonsense and three early frameshift variants of TBC1D2B were reported in affected individuals [1, 2]. In subject fibroblasts with the homozygous p.(Leu793*) variant and the compound heterozygous variants p.(Leu220Glufs*6) and p.(Tyr765*), TBC1D2B mRNA levels were drastically reduced to 10–20% compared to control cells and the TBC1D2B protein was absent. In a TBC1D2B cellular knockout model, reduced epidermal growth factor internalization and enhanced susceptibility to cell death under serum starvation condition were detected. The data led us to suggest that combined defects in vesicle trafficking and cell survival may underlie the neurodegenerative phenotype in subjects with TBC1D2B deficiency [1].
Here we report on five new subjects with novel biallelic TBC1D2B pathogenic variants. We investigated the impact of TBC1D2B variants on transcript and protein levels in leukocytes or fibroblasts from two subjects. Our findings support the loss-of-function nature of TBC1D2B variants. To describe the clinical spectrum of loss of TBC1D2B, we report genetic variants and detailed clinical phenotypic data of 10 total cases, including five newly reported cases, two brothers with a homozygous TBC1D2B frameshift variant [2], and the updated phenotypic data from the original cohort of three subjects with homozygous or compound heterozygous TBC1D2B variants [1]. Our findings define the core phenotypic traits associated with TBC1D2B loss-of-function, establishing a distinct and recognizable genetic syndrome.
Subjects and methods
Subjects
Informed consent for genetic analyses was obtained for subjects 7–11. Genetic studies were performed clinically or as approved by local Institutional Review Boards at University College London Hospitals (London, United Kingdom), Erciyes University (Kayseri, Turkey), Cincinnati Children’s Hospital (Cincinnati, USA), and Newfoundland and Labrador Health Services (Newfoundland and Labrador, Canada). The parents of the subjects provided written informed consent for participation in the study, clinical data and specimen collection, genetic analysis, and publication of relevant findings. Written informed consent for the publication of photographs was obtained for five subjects. Subject 10 and parents were enrolled in the Congenital Dyserythropoietic Anemia (CDA) Registry of North America (CDAR; clinicaltrials.gov NCT02964494), since the patient was diagnosed with pancytopenia and dyserythropoiesis.
Exome or genome sequencing and variant validation and segregation
Genomic DNA was extracted from peripheral blood samples using standard procedures. Sequencing was performed by either trio exome sequencing (subjects 7 and 11 with their respective parents), trio whole genome sequencing (subject 10 and parents), or proband-only exome sequencing (subject 8) (Supplementary information). When necessary, TBC1D2B variants were validated and segregated by Sanger-sequencing using either fibroblast-derived DNA (subject 7) or leukocyte-derived DNA (subjects 8 and 9 and parents) (Supplementary information). Primer sequences can be found in Supplementary Table 1.
Cell culture of primary dermal fibroblasts
Primary fibroblasts were cultured as described (Supplementary information) [1].
RNA isolation, cDNA synthesis, RT-PCR and Sanger-sequencing, and quantitative real-time PCR (RT-qPCR)
To analyze TBC1D2B transcripts, RNA isolation from fibroblasts, complementary DNA (cDNA) synthesis, reverse transcription polymerase chain reaction (RT-PCR), and Sanger-sequencing of amplicons to analyze TBC1D2B transcripts were performed as described [9]. RT-qPCR was performed to determine the relative mRNA levels of TBC1D2B as described (Supplementary information) [9]. Primer sequences can be found in Supplementary Table 1.
Immunoblotting
Whole-cell lysates from subject and control fibroblasts were prepared and immunoblotting was performed as described (Supplementary information) [1]. The antibodies used are described in Supplementary information.
Data analysis and statistics
Quantitative data are presented by GraphPad Prism 8 software (Instat, GraphPad Software) as the mean ± standard deviation (SD). For quantification, one- or two-way ANOVA followed by a Dunnett post hoc test for multiple comparisons was performed. A P < 0.05 was considered statistically significant (**P ≤ 0.01; ***P ≤ 0.001).
Results
Molecular genetic investigations
Since the first report on TBC1D2B as a novel disease gene for an autosomal recessive neurological disorder, we recruited five new patients (subjects 7 to 11) with biallelic TBC1D2B variants through GeneMatcher [10] or referral, including three unrelated subjects (subjects 7, 10, and 11) and a female patient and her brother (subjects 8 and 9) (Table 1).
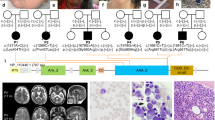
Trio exome sequencing identified compound heterozygous TBC1D2B (NM_144572.2) variants c.2584C>T; p.(Arg862Cys) and c.2758C>T; p.(Arg920*) in subject 7 (Fig. 1, Table 1 and Supplementary Table 2). By single exome sequencing, the 18-year-old female subject 8 was found to carry the homozygous transition c.360+1G>T affecting the highly conserved splice donor site in intron 1 of TBC1D2B (Fig. 1, Table 1 and Supplementary Table 2). Segregation analysis revealed the same homozygous TBC1D2B splice site variant in her similarly affected brother (subject 9) (Table 1 and Supplementary Fig. 1) and both healthy parents were heterozygous carriers of the variant (Supplementary Fig. 1). Trio genome sequencing in subject 10 and parents detected compound heterozygous TBC1D2B nonsense variants c.159C>G; p.(Tyr53*) and c.2353C>T; p.(Arg785*) in subject 10 (Fig. 1, Table 1, Supplementary Fig. 2 and Supplementary Table 2). In subject 11, the homozygous TBC1D2B nonsense variant c.25G>T; p.(Glu9*) was identified by trio exome sequencing (Fig. 1, Table 1 and Supplementary Table 2). The TBC1D2B variants p.(Arg862Cys) and c.360+1G>T are absent in the gnomAD database (v4.0.0), while p.(Glu9*) has a minor allele frequency of 0.001198%, p.(Tyr53*) of 0.000522%, p.(Arg785*) of 0.000138%, and p.(Arg920*) of 0.000518% in the gnomAD database v4.0.0 (Supplementary Table 2). The four nonsense variants are likely loss-of-function alleles, but because p.(Arg920*) is located in the last exon of TBC1D2B, it potentially escapes nonsense-mediated mRNA decay (NMD). The missense variant c.2584C>T; p.(Arg862Cys) is predicted to have a deleterious impact on protein function by several in silico tools (Supplementary Table 2). Arginine 862 is located in the RABGAP domain (amino acids 659–879) of TBC1D2B (NP_653173.1; Fig. 1) and shows evolutionary conservation between species (Supplementary Fig. 3). According to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [11], we interpreted TBC1D2B variants c.25G>T; p.(Glu9*) (PVS1, PP4_strong, PM2_supporting, and PM3_supporting), c.159C>G; p.(Tyr53*), c.2353C>T; p.(Arg785*) (for both: PVS1, PP4_strong, PM2_supporting, and PM3), c.2758C>T/p.(Arg920*) (PVS1_moderate, PP4_strong, PM2_supporting, PM3, and PS3), and c.360+1G>T (PVS1_RNA, PM2_supporting, PM3, and PP4_strong) as pathogenic variants, whereas the missense variant c.2584C>T; p.(Arg862Cys) (PP4_strong, PP3_supporting, PM2_supporting, and PM3) was interpreted as likely pathogenic variant. We, therefore, believe that the biallelic TBC1D2B variants likely explain the phenotype in subjects 7 to 11.

Exon-intron structure of the TBC1D2B gene (top) showing the locations of the pathogenic variants. Exons are indicated by boxes and introns by lines. The 5′ untranslated region is depicted in light gray, the 3′ untranslated region in white and the coding region in dark gray. Exon numbering and variant description are given according to the mRNA reference number NM_144572.2. The TBC1D2B domain structure (bottom) with amino acids (aa) numbered according to NP_653173.1. The position of the amino acid residue arginine 862 substituted by cysteine [p.(Arg862Cys)] is highlighted by a black line. Domain assignment was determined using the Simple Modular Architecture Research Tool (SMART) server, available at http://smart.embl-heidelberg.de/. CC coiled coil domain, LIR LC3-interacting region (aa 252-EEWELLD-258), PH pleckstrin homology domain, RABGAP RABGTPase-activating protein domain.
TBC1D2B transcript and protein analyses
We used fibroblasts of subject 7 to analyze the effect of the TBC1D2B variants p.(Arg862Cys) and p.(Arg920*) on RNA and protein level. We first confirmed the presence of both TBC1D2B variants in genomic DNA isolated from subject 7-derived fibroblasts (Fig. 2A). Next, we used qualitative RT-PCR to amplify TBC1D2B transcripts harboring the missense variant c.2584C>T; p.(Arg862Cys) and the nonsense variant c.2758C>T; p.(Arg920*). Sequence analysis of the amplicons revealed a ~1:1-ratio of both TBC1D2B mutant transcripts suggesting NMD escape of TBC1D2B mRNAs with the premature stop codon in fibroblasts of subject 7 (Fig. 2A). This was confirmed by quantitative real-time PCR as we detected similar TBC1D2B mRNA levels in subject 7 and control fibroblasts by using two different primer sets (Fig. 2B). We next determined TBC1D2B protein levels by immunoblotting and detected a statistically significant reduced level by ~50% in subject 7 compared to control cells (Fig. 2C, D). A C-terminally truncated TBC1D2B protein [p.(Arg920*)] with a predicted molecular mass of ~105 kDa (compared to wild-type TBC1D2B with ~110 kDa) could not be detected in cells from subject 7 (Fig. 2C), although we used an anti-TBC1D2B antibody that recognizes the central amino acids 372–452 (epitope). These data suggest that the TBC1D2B-Arg862Cys mutant protein seems to be stable, while the TBC1D2B mutant with loss of the last 44 amino acid residues at the C-terminus [p.(Arg920*)] is potentially unstable and/or degraded in fibroblasts of subject 7.

A Validation of the TBC1D2B c.2584C>T and c.2758C>T variants in DNA and transcript analysis using cDNA isolated from subject 7-derived fibroblasts. Partial sequence electropherograms showing the TBC1D2B c.2584C>T; p.(Arg862Cys) and c.2758C>T; p.(Arg920*) variants in fibroblast-derived DNA of subject 7 in the heterozygous state (left column). The right column shows sequence electropherograms of TBC1D2B transcripts obtained from fibroblast-derived cDNA of subject 7. The ratio of both mutant TBC1D2B transcripts is ~1:1. Arrows point to the position of the pathogenic variant. B Relative TBC1D2B mRNA levels determined by real-time quantitative PCR (RT-qPCR) using two primer pairs (exon 8–9 and exon 10–11) with fibroblast-derived cDNA of subject 7 and three controls normalized to GAPDH mRNA levels. The mean ± SD of one experiment performed in triplicate is shown. C Immunoblot analysis of lysates obtained from fibroblasts of subject 7 and three controls from three different passages. The amount of TBC1D2B was monitored by using an anti-TBC1D2B antibody. An anti-Tubulin antibody was used to control for equal loading. D Band intensities were quantified using a chemiluminescence imager. TBC1D2B was normalized relative to Tubulin. The mean ± SD of three independent experiments is shown. One-way ANOVA followed by Dunnett post-hoc test: **P ≤ 0.01. cDNA complementary DNA, Ctrl. 1–3 fibroblast controls, ns not significant.
We next investigated the effect of the homozygous c.360+1G>T change in intron 1 on TBC1D2B pre-mRNA splicing by using leukocyte-derived RNA from subject 8. We used a primer pair with the forward primer located in exon 1 and the reverse primer in exon 3 in RT-PCR experiments (Supplementary Table 1). We generated an RT-PCR product in subject 8 that was about 35 bp smaller than the wild-type amplicons generated from control-derived cDNAs (Fig. 3A). Direct sequencing of the subject 8-derived amplicon revealed TBC1D2B mutant transcripts with loss of the last 33 bp of exon 1 (r.328_360del) due to activation of a cryptic splice donor site in exon 1 (Fig. 3B). TBC1D2B r.328_360del mRNAs could lead to potential production of a TBC1D2B mutant protein which lacks 11 amino acid residues from valine 110 to lysine 120 [p.(Val110_Lys120del)] (Fig. 3B). Quantitative real-time PCR identified a statistically significantly reduced TBC1D2B mRNA level to 15–21% in leukocytes of subject 8 compared with controls (Fig. 3C and Supplementary Fig. 4). Drastic decrease of TBC1D2B total mRNA level in leukocytes from subject 8 suggests expression of other aberrantly spliced TBC1D2B transcripts in addition to TBC1D2B r.328_360del mRNAs from both TBC1D2B c.360+1G>T alleles. These other aberrantly spliced TBC1D2B mRNAs may be efficiently degraded by NMD in subject 8-derived leukocytes, as they cannot be detected by qualitative and quantitative RT-PCR analyses. Potential production of a TBC1D2B p.(Val110_Lys120del) mutant protein could not be analyzed due to lack of patient-derived fibroblasts from subject 8.

A Agarose gel showing RT-PCR products with a primer pair for TBC1D2B exons 1–3 using leukocyte-derived cDNA of subject 8 and two controls. Schematics of the exon-exon junctions and the size of the amplicons (after sequencing) are depicted on the right. A single amplicon of ~250 bp was generated by RT-PCR in subject 8 and of ~290 bp in controls. B Partial sequence electropherograms obtained from Sanger-sequencing of RT-PCR amplicons of subject 8 and control 4. Exon numbering is given. Dotted lines mark the junction between exons 1 and 2. A cryptic splice donor site 33 bp upstream of the canonical splice donor site (red rectangle) is used in leukocytes of subject 8 due to the homozygous TBC1D2B c.360+1G>T intron 1 variant. The resulting TBC1D2B transcripts lack 33 bp of exon 1 (Δ33 bp; r.328_360del), which is predicted to cause an in-frame loss of 11 amino acids on protein level [p.(Val110_Lys120del)]. C Relative TBC1D2B mRNA levels determined by real-time quantitative PCR (RT-qPCR) using two primer pairs (exon 8–9 and exon 10–11) with leukocyte-derived cDNA of subject 8 and three controls normalized to GAPDH mRNA levels. Two independent RNA isolations (Experiment 1 and 2) of subject 8 were used against the same set of controls. The mean ± SD of two experiments each performed in triplicate is shown. Results of individual experiments are shown in Supplementary Fig. 4. Two-way ANOVA followed by Dunnett post-hoc test: **P ≤ 0.01, ***P ≤ 0.001. bp base pair, cDNA complementary DNA, Ctrl. 4–6 leukocyte controls, Ex exon, H2O water control of the RT-PCR.
Clinical findings in ten subjects
In Table 1, we briefly summarized clinical and neuroimaging features of all ten subjects carrying biallelic TBC1D2B variants known to date. These include new follow-up data from subjects 1, 3, and 4 [1], previously published data from subjects 5 and 6 [2], and data from subjects 7 to 11 identified in this study. Clinical case reports of the five new patients and comprehensive clinical data of the ten subjects can be found in Supplementary information and Supplementary table 3. We here summarize commonly observed clinical features of subjects 1 and 3 to 11.
Abnormality of the face
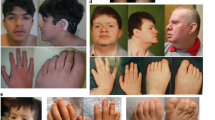
Five subjects had coarse facial features (Fig. 4A). Eight subjects had gingival overgrowth that started early in childhood and required surgery in a few individuals. An abnormal mandible morphology was found in eight subjects. Five subjects had a prominent mandible (Fig. 4A), while three subjects had been diagnosed with fibrous dysplasia of the mandibular bones (Fig. 4B and [2]). In two subjects (brothers), soft tissue growth also involved the maxilla region [2]. Cherubism had been diagnosed in two subjects [1, 2]. Pigmented freckles appeared on the face of two subjects in adulthood.

A We assessed the facial features of subjects for whom photographs were available and for whom authorization has been obtained for disclosure. From left to right: facial photographs of subject 1 (S1) at age 31 years, subject 7 (S7) at 4 years (after gingival hyperplasia surgery), 23 years, and 26 years (a few days before she died), subject 8 (S8) at 10 years, subject 9 (S9), subject 8’s brother, at 29 years, and subject 11 (S11) at 34 years. All subjects except subject 11 had mandibular prognathia. Subject 1 had a coarse face with prominent nose, low hanging columella, and pigmented freckles on face. The face of subject 7 changed over time and the mandible became more prominent. At the age of 23 years, subject 7 had brown freckles on her face. Subject 8 had facial asymmetry and a low hanging columella. Low hanging columella was also present in subject 9. Subject 11 had mild hypertelorism, long palpebral fissures, mild eversion of the lateral lower eyelids, short nose with anteverted nares, smooth philtrum, and thin upper lip vermilion. She also had a mildly prominent premaxillary region. She presented with a malar rash and bilateral infraorbital xanthelasma. B Orthopantogram of subject 3 (S3) done at age 11 years shows extensive fibrous dysplasia of the mandible, which is stable when compared to previous orthopantogram done at age 8.5 years [compare with Fig. 1c published in Harms et al.] [1], (C) Finger and toe abnormalities in subjects 1 and 9. Photographs of hands and feet of subject 1 show flexion contractures of fingers and toes at the age of 31 years. Photographs of left hand and left foot of subject 9 at the age of 29 years show flexion contractures of fingers and tibial deviation of the 4th toe. S, subject; y, years.
Abnormality of the nervous system
Five subjects had a normal development in childhood, while developmental delay was present in the other five subjects. Mental deterioration or cognitive regression became apparent in six subjects between the ages of 5 and 20 years and was associated with gait ataxia and slurred speech. Two subjects had no speech development, and two other subjects were unable to speak in their twenties and thirties. Eight subjects developed seizures between the ages of 4 months and 32 years that were controlled with medication in four. Five subjects showed behavioral abnormalities. Brain abnormalities included cerebellar and/or cerebral atrophy in six subjects and lateral ventricle dilatation in three subjects. Movement abnormalities were reported in six subjects, including tremors, dystonia, and myoclonus. One subject could not walk as a toddler, and two adult subjects could walk with assistance. Three subjects became bedridden between the ages of 12 and 36 years and had respiratory failure requiring assisted ventilation.
Abnormality of the eye
Visual loss was noted in five subjects between the ages of 3 and 20 years. One subject had complex ophthalmologic abnormalities, in part due to pre-maturity. Macular optical coherence tomography revealed inner layer schisis-like cavitation as well as vacuoles and deposition in ganglion cell layer in one subject.
Additional abnormalities
Two subjects had bilateral hearing loss, and one had minor hearing issues. Seven subjects had flexion contractures involving fingers, toes, elbows, knees, arms, and/or legs (Fig. 4C), which began early in two, at 3–5 years of age. Five subjects had low blood cell counts, with two subjects with pancytopenia and one subject each diagnosed with thrombocytopenia, leukopenia or mild lymphopenia and thrombocytopenia. Seven subjects were alive; the oldest were 31 and 35 years old. Three subjects died between the ages of 21 and 39 years.
Discussion
With the five additionally described subjects with biallelic TBC1D2B variants, 12 different disease-associated TBC1D2B variants have now been reported, including seven nonsense, three frameshift, one splice site, and one missense variant (Supplementary Table 2). The nonsense, frameshift, and splice site variants likely represent loss-of-function alleles and lead to TBC1D2B deficiency when present in a homozygous or compound heterozygous state in subject fibroblasts [1]. In fibroblasts of subject 7 with compound heterozygous TBC1D2B variants c.2758C>T; p.(Arg920*) and c.2584C>T; p.(Arg862Cys), normal TBC1D2B mRNA level as well as TBC1D2B protein level of about 50% compared with control cells were detected. Any C-terminally truncated TBC1D2B protein that may be produced is likely to be unstable and degraded. In contrast, the data indicate the TBC1D2B-Arg862Cys mutant protein is normally produced and stable, and may have an impaired GAP activity. In leukocytes of subject 8 with the homozygous c.360+1G>T variant, aberrantly spliced TBC1D2B mRNAs in addition to drastically reduced TBC1D2B mRNA level were detected. Together, the disease-associated TBC1D2B alleles identified up to date likely lead to complete loss of TBC1D2B or significantly impaired TBC1D2B function.
By analyzing clinical features of ten subjects with biallelic TBC1D2B loss-of-function variants we delineated a core phenotype. Cognitive and motor development of affected individuals can be normal or delayed, and most of the subjects develop seizures. Gingival overgrowth usually starts in early childhood. Mandibular prognathia is a common feature, with fibrous dysplasia of the mandible and/or maxilla in a few subjects. Progressive neurological deterioration was observed and begins between the ages of 10 and 20. Mental deterioration can be severe, and death can occur in early adulthood. Flexion contracture is a common feature. Visual loss and low blood cell counts have been observed in half of the subjects. Together, the TBC1D2B disorder is a progressive neurological disease with gingival overgrowth and mandibular prognathia or cherubism as characteristic associated abnormalities. Hematological abnormality has not yet been reported and seems to be a novel feature associated with TBC1D2B deficiency. The combination of manifestations makes TBC1D2B disorder a clinical recognizable phenotype, at least in subjects who have developed progressive disease with neurological deterioration. Although all TBC1D2B pathogenic variants reported to date likely are loss-of-function variants, the phenotypic presentation in the ten subjects is variable. For example, the 35-year-old subject 11 with the homozygous early stop variant p.(Glu9*) had a relatively mild phenotype, whereas subject 7 with a nonsense and a missense variant in trans presented with severe mental deterioration and decline and died at the age of 26 years and 10 months. In the literature, a 21-year-old man and an 11-year-old girl with clinical features overlapping those of the TBC1D2B disorder have been reported, including gingival overgrowth since early childhood, osteofibrosis of the maxillary alveolar bone or cherubism, mental or psychomotor retardation, and cerebral and/or cerebellar atrophy. Chromosome analysis revealed a normal karyotype in both subjects [12, 13]. Genetic analysis of the TBC1D2B gene would be valuable in the two subjects to potentially identify additional individuals with this rare disorder.
Fibrous dysplasia of the mandible and/or maxilla was observed in three subjects and diagnosed as cherubism in two (Supplementary Table 3). Cherubism is characterized by bilateral proliferative fibro-osseous lesions limited to the mandible and maxilla. Usually, this is an isolated childhood-onset, self-limited bone disease without other physical abnormalities [14]. Isolated cherubism is an autosomal dominant condition and caused by heterozygous missense variants in SH3BP2, mainly affecting codons 415 and 418–420 and causing a gain-of-function effect [15,16,17,18]. SH3BP2 is a cytoplasmic adapter protein and binds to a range of proteins, including CIN85 and HIP-55 that are involved in endocytic and cytoskeletal regulation [19]. We hypothesize that a possible function of SH3BP2 in intracellular trafficking may overlap with the proposed role of TBC1D2B in membrane fusion and vesicle trafficking [1, 8].
Clinical features of subjects with biallelic TBC1D2B pathogenic variants show phenotypic overlap with syndromic neurodevelopmental K+ channelopathies or Zimmermann-Laband syndrome associated with dominant KCNN3, KCNH1, and KCNK4 variants. Affected individuals present with developmental delay and/or intellectual disability, coarse facial features, gingival overgrowth, hypoplastic nails and/or distal phalanges, and hypertrichosis [20]. However, subjects with biallelic TBC1D2B variants did not have hypertrichosis and hypoplastic distal phalanges of fingers and/or toes. Ramon syndrome, another rare condition, characterized by intellectual disability, seizures, cherubism, gingival overgrowth, hypertrichosis, and short stature [21,22,23,24,25,26,27,28], also shows clinical overlap with the subjects reported here. While a hallmark of the TBC1D2B disorder is mental deterioration associated with cerebellar and/or cerebral atrophy and lateral ventricular dilatation, developmental or cognitive regression has not been described in Ramon syndrome [22, 26, 28].
With the recent data that TBC1D2B positively regulates autophagy [29], an imbalanced crosstalk between autophagy and the endolysosomal pathway can be assumed in subjects with biallelic TBC1D2B variants [30]. Importantly, defects in autophagy and the endolysosomal system have been associated with neuronal dysfunction and neurodegenerative diseases [31, 32], raising the possibility that the TBC1D2B-related disease belongs to this group of disorders.
Data availability
The data that supports the findings of this study are available within the paper and in the supplementary information. Genome and exome sequencing data are not publicly available due to privacy or ethical restrictions. The novel TBC1D2B variants reported in subjects 7–11 in this manuscript were submitted to the LOVD database (https://databases.lovd.nl/shared/genes/TBC1D2B), with the LOVD variant IDs: #0000927486, #0000927487, #0000927488, #0000927489, #0000952085, #0000952086, and #0000952087.
References
Harms FL, Parthasarathy P, Zorndt D, Alawi M, Fuchs S, Halliday BJ, et al. Biallelic loss-of-function variants in TBC1D2B cause a neurodevelopmental disorder with seizures and gingival overgrowth. Hum Mutat. 2020;41:1645–61.
Correia-Costa GR, de Leeuw N, Pfundt R, Sgardioli IC, Dos Santos AP, de Lima Santos M, et al. Biallelic frameshift variant in the TBC1D2B gene in two siblings with progressive gingival overgrowth, fibrous dysplasia of face, and mental deterioration. Clin Genet. 2022;102:537–42.
Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim Biophys Acta. 2003;1603:47–82.
Fukuda M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci Rep. 2011;31:159–68.
Borchers AC, Langemeyer L, Ungermann C. Who’s in control? Principles of Rab GTPase activation in endolysosomal membrane trafficking and beyond. J Cell Biol. 2021;220:e202105120.
Kanno E, Ishibashi K, Kobayashi H, Matsui T, Ohbayashi N, Fukuda M. Comprehensive screening for novel rab-binding proteins by GST pull-down assay using 60 different mammalian Rabs. Traffic. 2010;11:491–507.
Munro S, Gillingham AK, Bertram J, Begum F. In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation. Elife. 2019;8:e45916.
Wei D, Zhan W, Gao Y, Huang L, Gong R, Wang W, et al. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 2021;31:157–77.
Schneeberger PE, Kortum F, Korenke GC, Alawi M, Santer R, Woidy M, et al. Biallelic MADD variants cause a phenotypic spectrum ranging from developmental delay to a multisystem disorder. Brain. 2020;143:2437–53.
Sobreira N, Schiettecatte F, Boehm C, Valle D, Hamosh A. New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum Mutat. 2015;36:425–31.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
He L, Ping FY. Gingival fibromatosis with multiple unusual findings: report of a rare case. Int J Oral Sci. 2012;4:221–5.
Yalcin S, Yalcin F, Soydinc M, Palanduz S, Gunhan O. Gingival fibromatosis combined with cherubism and psychomotor retardation: a rare syndrome. J Periodontol. 1999;70:201–4.
Kannu P, Baskin B, Bowdin S Cherubism. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 1993-2024.
Chrcanovic BR, Guimaraes LM, Gomes CC, Gomez RS. Cherubism: a systematic literature review of clinical and molecular aspects. Int J Oral Maxillofac Surg. 2021;50:43–53.
Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125–6.
Lietman SA, Yin L, Levine MA. SH3BP2 mutations potentiate osteoclastogenesis via PLCgamma. J Orthop Res. 2010;28:1425–30.
Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, et al. Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell. 2007;128:71–83.
Le Bras S, Moon C, Foucault I, Breittmayer JP, Deckert M. Abl-SH3 binding protein 2, 3BP2, interacts with CIN85 and HIP-55. FEBS Lett. 2007;581:967–74.
Gripp KW, Smithson SF, Scurr IJ, Baptista J, Majumdar A, Pierre G, et al. Syndromic disorders caused by gain-of-function variants in KCNH1, KCNK4, and KCNN3-a subgroup of K(+) channelopathies. Eur J Hum Genet. 2021;29:1384–95.
Mehawej C, Hoischen A, Farah RA, Marey I, David M, Stora S, et al. Homozygous mutation in ELMO2 may cause Ramon syndrome. Clin Genet. 2018;93:703–6.
Ramon Y, Berman W, Bubis JJ. Gingival fibromatosis combined with cherubism. Oral Surg Oral Med Oral Pathol. 1967;24:435–48.
Surej Kumar LK, Deepa DS, Dilna S. Ramon Syndrome- A Rare Form of Cherubism. Ann Maxillofac Surg. 2019;9:415–8.
de Pina-Neto JM, de Souza NV, Velludo MA, Perosa GB, de Freitas MM, Colafemina JF. Retinal changes and tumorigenesis in Ramon syndrome: follow-up of a Brazilian family. Am J Med Genet. 1998;77:43–6.
Parkin B, Law C. Axenfeld anomaly and retinal changes in Ramon syndrome: follow-up of two sibs. Am J Med Genet. 2001;104:131–4.
Pina-Neto JM, Moreno AF, Silva LR, Velludo MA, Petean EB, Ribeiro MV, et al. Cherubism, gingival fibromatosis, epilepsy, and mental deficiency (Ramon syndrome) with juvenile rheumatoid arthritis. Am J Med Genet. 1986;25:433–41.
Pridmore C, Baraitser M, Leonard J. Ramon syndrome with diabetes mellitus and vascular skin lesions in two sibs. Clin Dysmorphol. 1992;1:29–35.
Suhanya J, Aggarwal C, Mohideen K, Jayachandran S, Ponniah I. Cherubism combined with epilepsy, mental retardation and gingival fibromatosis (Ramon syndrome): a case report. Head Neck Pathol. 2010;4:126–31.
Hoffmann ME, Jacomin AC, Popovic D, Kalina D, Covarrubias-Pinto A, Dikic I. TBC1D2B undergoes phase separation and mediates autophagy initiation. J Cell Biochem. 2024; https://doi.org/10.1002/jcb.30481.
Birgisdottir AB, Johansen T. Autophagy and endocytosis—interconnections and interdependencies. J Cell Sci. 2020;133:jcs228114.
Giovedi S, Ravanelli MM, Parisi B, Bettegazzi B, Guarnieri FC. Dysfunctional autophagy and endolysosomal system in neurodegenerative diseases: relevance and therapeutic options. Front Cell Neurosci. 2020;14:602116.
Papandreou ME, Tavernarakis N. Crosstalk between endo/exocytosis and autophagy in health and disease. Biotechnol J. 2020;15:e1900267.
Acknowledgements
We thank all subjects and families for their participation in this study and Jane Rehberg, Valentin Keil, and Henrike Wilshusen for their skillful technical assistance. We thank Ammar Husami, the bioinformatician of the CDA Registry in Cincinnati Children’s Hospital Medical Center, for the analysis of the genome sequencing trio for subject 10. We acknowledge the Clinical Neurogenomics Research Center (CNRC) in the Department of Neurology of the David Geffen School of Medicine at UCLA for assistance with sample collection and biobanking.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (KU 1240/13-1 to KK), NIH (K08 NS105916 to JER), the Wellcome Trust, and the MRC. We acknowledge financial support from the Open Access Publication Fund of UKE - Universitätsklinikum Hamburg-Eppendorf and DFG – German Research Foundation. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: Frederike L. Harms, Kerstin Kutsche; Formal analysis and investigation: Frederike L. Harms, Jessica Erin Rexach, Stephanie Efthymiou, Busra Aynekin, Hüseyin Per, Ayten Güleç, Sheela Nampoothiri, Hugo Sampaio, Rani Sachdev, Radka Stoeva, Kasiani Myers, Loren D. M. Pena, Theodosia A. Kalfa, Marisa Chard, Megan Klassen, Megan Pries; Writing—original draft preparation: Frederike L. Harms, Jessica Erin Rexach, Stephanie Efthymiou, Busra Aynekin, Hüseyin Per, Ayten Güleç, Sheela Nampoothiri, Hugo Sampaio, Rani Sachdev, Radka Stoeva, Kasiani Myers, Loren D. M. Pena, Theodosia A. Kalfa, Marisa Chard, Megan Klassen, Megan Pries, Kerstin Kutsche; Writing—review and editing: Frederike L. Harms, Jessica Erin Rexach, Stephanie Efthymiou, Kerstin Kutsche; Funding acquisition: Jessica Erin Rexach, Stephanie Efthymiou, Kerstin Kutsche; Supervision: Kerstin Kutsche. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Genetic studies were performed clinically (as approved by the Provincial Medical Genetics Program at Newfoundland and Labrador Health Services) or as approved by the local Institutional Review Board at University College London Hospitals (IRAS: 310045; REC: 22/NE/0080; London, United Kingdom), the Clinical Research Ethics Committee of the Erciyes University (2015/226; Kayseri, Turkey), and the Cincinnati Children’s Hospital Medical Center Institutional Review Board (CCHMC IRB# 2016–2727). Written informed consent for the publication of photographs was obtained for five subjects.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harms, F.L., Rexach, J.E., Efthymiou, S. et al. Loss of TBC1D2B causes a progressive neurological disorder with gingival overgrowth. Eur J Hum Genet (2024). https://doi.org/10.1038/s41431-024-01563-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-024-01563-5
