
Abstract
Wilms tumour (nephroblastoma) is a renal embryonal tumour that is frequently caused by constitutional variants in a small range of cancer predisposition genes. TRIM28 has recently been identified as one such gene. Previously, observational data strongly suggested a parent of origin effect, whereby Wilms tumour only occurred following maternal inheritance of a pathogenic genetic variant. However, here we report a child with bilateral Wilms tumour who had inherited a pathogenic TRIM28 variant from their father. This finding suggests that genetic counselling for paternally inherited pathogenic variants in TRIM28 should include discussion of a potential risk of Wilms tumour.
Similar content being viewed by others

Introduction
Wilms tumour (nephroblastoma) is an embryonal tumour of kidney tissue that is the most frequent renal neoplasm in childhood. It results from abnormal nephrogenesis and is associated with aberrant mesenchymal to epithelial transition [1]. Most Wilms tumours occur as isolated cases and recent evidence has demonstrated that in some instances, post-zygotic genomic events early in embryogenesis lead to pre-malignant clonal expansions that are widespread in kidney tissue from affected individuals [2].
Additionally, there are a number of inherited causes that occur due to constitutional pathogenic variants (e.g. in WT1, DIS3L2, REST, BUB1B etc) [3] and Wilms tumour predisposition is a well-recognised feature of the congenital imprinting disorder Beckwith-Wiedemann Syndrome [4]. Recently, TRIM28 has been described independently as a further Wilms predisposition gene by multiple groups and appears particularly associated with the rarer epithelial histological subtype along with loss of the wild type allele in tumour [5,6,7,8,9,10,11]. TRIM28 is located in the terminal part of the long arm of chromosome 19 and encodes a transcriptional co-repressor. It has been implicated in a variety of cellular processes that are potentially relevant to Wilms tumour including epithelial to mesenchymal transition, DNA damage response, stem cell maintenance [12], and kidney development [13].
A striking feature of reported Wilms tumour cases associated with constitutional TRIM28 variants is an apparent parent of origin effect whereby the neoplasm only manifests following maternal inheritance of the genetic variant. Such parent-of-origin effects are a characteristic of pathogenic variants in genes which undergo genomic imprinting (i.e. are expressed from only one parental allele in a parent-specific manner) [14]. However, parent-of-origin effects on tumour risks have also been described in other tumour predisposition syndromes caused by variants in non-imprinted genes (e.g. phaeochromocytoma/paraganglioma (PPGL) due to constitutional SDHD and MAX variants) when the predisposition gene maps to a chromosome (e.g. 11 and 14) that contain an imprinted gene cluster [15, 16]. Here, we report the first case of Wilms tumour due to documented paternal transmission of a TRIM28 pathogenic variant.
Subjects and methods
The family was ascertained via referral to the Department of Clinical Genetics, Cambridge University Hospitals. Previous research conducted by the Institute of Cancer Research as part of the Factors Associated with Childhood Tumours (FACT) study had identified the TRIM28 variant, which was confirmed in a diagnostic laboratory. Consent for further study was through participation in the (Molecular Pathology of Human Genetic Disease study) and written informed consent for publication was obtained. Molecular analysis of tumour tissue was attempted by preparing sections from formalin fixed paraffin embedded (FFPE) biopsy samples from the left and right kidneys. However, DNA was not of sufficient quality for reliable sequencing.
Results
Case report
A female proband presented with abdominal distension and hypertension at the age of nine months and was diagnosed with bilateral Wilms tumour (with two masses on the left), which was treated with chemotherapy and nephron sparing surgery. Histological examination of both left and right tumour biopsies showed predominantly epithelial structures and some blastemal areas with positive immunostaining for WT-1, INI-1 and CD56. Nephrogenic rests were considered likely to be present.
There was no known family history of cancer other than a lymphoma diagnosis in the paternal grandfather.
Molecular studies
Genetic investigation of blood DNA with chromosomal microarray and WT1 testing did not demonstrate any potentially causative variants and the patient was recruited to a research study for further consideration of a genetic cause. This demonstrated a constitutional TRIM28 nonsense variant (NM_005762 c.688 C > T p.(Arg230Ter)) via exome sequencing that was deemed pathogenic and causative of the Wilms tumours. Subsequent testing of parental samples for the variant showed that it had been unexpectedly inherited from the father, who had no significant medical history at age 36 years. No fertility treatment was known to have been necessary for the pregnancy and the couple have two other children. No other Wilms tumours had occurred in the family.
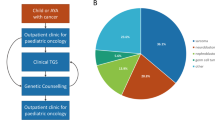
The proband continued under oncology follow up with ultrasound scans every three months until age seven years with six monthly scans thereafter. Three monthly scans were recommended until age seven years for other carriers of the paternally inherited variant in the family. Individuals with the variant who were above the age for Wilms tumour surveillance [3] had a single one-off scan, which showed two likely renal angiomyolipomas in the case of the father but no other abnormalities.
Discussion
The assertion that only maternal inheritance of TRIM28 variants predisposes to Wilms tumour arises from the observation that in one of the originally described series, 10/10 inherited (as opposed to de novo) variants were also present in the mother [5] and no instances of paternal inheritance were noted in other reports [6, 8, 9]. Such parent of origin effects in tumour predisposition syndromes have been described previously, notably for SDHD, SDHAF2 and MAX pathogenic variants, which are associated with PPGL when paternally inherited [15, 16].
Perhaps the most intuitive molecular explanation for parent of origin effects is that the allele from one parent is inactive in the normal state due to an imprint established during gametogenesis. In that scenario, inheritance of a pathogenic loss of function variant from that parent would not cause additional aberration of gene function but inheritance from the other parent would lead to two non-functional alleles. However, to date no evidence has been reported that TRIM28 is an imprinted gene [17, 18].
An alternative explanation of parent-of-origin effects associated with a non-imprinted cancer predisposition gene is that proposed for PPGL associated with pathogenic variants in SDHD and SDHAF2, which both map to chromosome 11 and predispose to PPGL when pathogenic variants are paternally inherited. In these cases, PPGL usually show loss of the maternally inherited chromosome 11 in the tumour tissue. This results in loss of the maternally expressed imprinted gene CDKN1C whilst the function of the paternally expressed growth factor IGF2 is unaffected [19]. In contrast when a germline SDHD/SDHAF2 variant is maternally inherited, though somatic loss of the paternal chromosome 11 would be associated with biallelic SDHD inactivation, there would also be loss of the functioning paternal IGF2 allele but the functional CDKN1C allele would be unaffected. Based on studies of PPGL associated with pathogenic variants in SDHD, it would be predicted that TRIM28 tumourigenesis would be favoured by somatic events that resulted in biallelic TRIM28 inactivation accompanied by loss of paternally expressed imprinted genes (and/or preservation of maternally expressed imprinted genes) mapping to chromosome 19. Penetrance from paternally inherited variants under this model would be due to mitotic recombination events involving this region (Fig. 1). Whilst poor DNA quality precluded investigation of FFPE- tumour material in the present case, other groups have found that in Wilms tumours associated with constitutional or somatic TRIM28 pathogenic variants, loss of heterozygosity (LOH) can involve imprinted genes on chromosome 19, albeit without complete consistency in terms of included genes. Halliday et al. reported LOH in a Wilms tumour from an individual with a constitutional TRIM28 frameshift variant where a distal q13.43 region of homozygosity included eight genes [6], none of which are firmly considered as imprinted [17]. Armstrong et al. performed copy number analysis in five Wilms tumours with somatic TRIM28 mutations, four of which showed copy number neutral LOH at 19q13.32 to 19q13.43 [7] that includes TRIM28 along with a number of reported paternally expressed imprinted genes (ZIM2, PEG3, MIMT1, MIR371A) [17, 18]. If the Halliday and Armstrong studies are taken together, no known imprinted genes are within the region of LOH in all tumours although some affected in multiple samples have greater potential relevance such as PEG3, a paternally expressed gene that was hypothesised as of potential relevance by Mahamdallie et al. [5]. It is a regulator of the tumour necrosis factor immune response and decreased expression has been associated with tumourigenesis [20].

Potential mechanism of penetrance resulting from maternal inheritance of TRIM28 variants and paternal inheritance in exceptional cases.
Another mechanism that has been proposed to explain the parent-of-origin effect is that TRIM28 pathogenic variants could affect spermatogenesis and lead to decreased male fertility. This has been supported by the observation of testicular degeneration and premature infertility in heterozygous TRIM28 knockout mice [21] in addition to some reported male pathogenic variant carriers not having fathered children [5, 8]. However, there was no indication of subfertility in the male carrier in this case.
This report of Wilms tumour in the context of a paternally inherited TRIM28 pathogenic variant demonstrates that this mode of inheritance is possible despite pre-existing evidence of a parent of origin effect where penetrance would only result from maternal inheritance. The mechanistic basis of this observation is unclear but more frequent paired WGS analysis of Wilms tumours in clinical practice may beget the definition of common regions of LOH and mitotic recombination events. Although influenced by ascertainment bias, the penetrance of maternally inherited TRIM28 pathogenic variants has been estimated at around 67% [5, 8, 9]. Whilst identification of a paternally inherited TRIM28 variant in a child appears to confer a lower risk, we suggest that clinicians still consider surveillance for Wilms tumour, particularly if it has been penetrant in a sibling. The question of surveillance is likely to arise with increasing frequency in the UK given that TRIM28 is one of only five cancer predisposition genes included in the Genomics England Newborn Genomes Programme [22].
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Rivera MN, Haber DA. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer. 2005;5:699–712. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16110318
Coorens THH, Treger TD, Al-Saadi R, Moore L, Tran MGB, Mitchell TJ, et al. Embryonal precursors of Wilms tumor. Sci [Internet]. 2019;366:1247–51. http://www.ncbi.nlm.nih.gov/pubmed/31806814
Hol JA, Jewell R, Chowdhury T, Duncan C, Nakata K, Oue T, et al. Wilms tumour surveillance in at-risk children: literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer [Internet]. 2021;153:51–63. http://www.ncbi.nlm.nih.gov/pubmed/34134020
Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol [Internet]. 2018;14:229–49. http://www.ncbi.nlm.nih.gov/pubmed/29377879
Mahamdallie S, Yost S, Poyastro-Pearson E, Holt E, Zachariou A, Seal S, et al. Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Heal. 2019;3:322–31.
Halliday BJ, Fukuzawa R, Markie DM, Grundy RG, Ludgate JL, Black MA, et al. Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet [Internet]. 2018;14:e1007399. http://www.ncbi.nlm.nih.gov/pubmed/29912901
Armstrong AE, Gadd S, Huff V, Gerhard DS, Dome JS, Perlman EJ. A unique subset of low-risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: a children’s oncology group study. PLoS One [Internet]. 2018;13:e0208936. http://www.ncbi.nlm.nih.gov/pubmed/30543698
Diets IJ, Hoyer J, Ekici AB, Popp B, Hoogerbrugge N, van Reijmersdal SV, et al. TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer [Internet]. 2019;145:941–51. http://www.ncbi.nlm.nih.gov/pubmed/30694527
Hol JA, Diets IJ, de Krijger RR, van den Heuvel-Eibrink MM, Jongmans MC, Kuiper RP. TRIM28 variants and Wilms’ tumour predisposition. J Pathol [Internet]. 2021;254:494–504. http://www.ncbi.nlm.nih.gov/pubmed/33565090
Moore C, Monforte H, Teer JK, Zhang Y, Yoder S, Brohl AS, et al. TRIM28 congenital predisposition to Wilms’ tumor: novel mutations and presentation in a sibling pair. Cold Spring Harb Mol case Stud. 2020;6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32699065
Wegert J, Fischer AK, Palhazi B, Treger TD, Hilgers C, Ziegler B, et al. TRIM28 inactivation in epithelial nephroblastoma is frequent and often associated with predisposing TRIM28 germline variants. J Pathol [Internet]. 2024;262:10–21. http://www.ncbi.nlm.nih.gov/pubmed/37792584
Czerwińska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci [Internet]. 2017;24:63. http://www.ncbi.nlm.nih.gov/pubmed/28851455
Dihazi GH, Jahn O, Tampe B, Zeisberg M, Müller C, Müller GA, et al. Proteomic analysis of embryonic kidney development: Heterochromatin proteins as epigenetic regulators of nephrogenesis. Sci Rep. [Internet]. 2015;5:13951. http://www.ncbi.nlm.nih.gov/pubmed/26359909
Eggermann T, Monk D, de Nanclares GP, Kagami M, Giabicani E, Riccio A, et al. Imprinting disorders. Nat Rev Dis Prim [Internet]. 2023;9:33. http://www.ncbi.nlm.nih.gov/pubmed/37386011
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Sci [Internet]. 2000;287:848–51. http://www.ncbi.nlm.nih.gov/pubmed/10657297
Comino-Méndez I, Gracia-aznárez FJ, Schiavi F, Landa I, Leandro-garcía LJ, Letón R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–7.
Ginjala V. Gene imprinting gateway. Genome Biol [Internet]. 2001;2:reports2009. https://genomebiology.biomedcentral.com/articles/10.1186/gb-2001-2-8-reports2009
Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet [Internet]. 2005;21:457–65. http://www.ncbi.nlm.nih.gov/pubmed/15990197
Hensen EF, Jordanova ES, van Minderhout IJHM, Hogendoorn PCW, Taschner PEM, van der Mey AGL, et al. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene [Internet]. 2004;23:4076–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15064708
Kohda T, Asai A, Kuroiwa Y, Kobayashi S, Aisaka K, Nagashima G, et al. Tumour suppressor activity of human imprinted gene PEG3 in a glioma cell line. Genes Cells [Internet]. 2001;6:237–47. http://doi.wiley.com/10.1046/j.1365-2443.2001.00412.x.
Tan JHL, Wollmann H, van Pelt AMM, Kaldis P, Messerschmidt DM. Infertility-causing haploinsufficiency reveals TRIM28/KAP1 requirement in spermatogonia. Stem Cell Rep. [Internet]. 2020;14:818–27. http://www.ncbi.nlm.nih.gov/pubmed/32302554
Genomics England. Genomics England Newborn Genomes Programme Conditions List [Internet]. 2023 [cited 2023 Nov 14]. Available from: https://www.genomicsengland.co.uk/initiatives/newborns/choosing-conditions/conditions-list-generation-study.
Acknowledgements
Tissue sample processing was provided by the Human Research Tissue Bank, Cambridge University Hospitals. DNA extraction and consultation surrounding sequencing was performed by the Stratified Medicine Core Laboratory, University of Cambridge (Ana Toribo and Graeme Clark).
Funding
This research was supported by the NIHR Cambridge Biomedical Research Centre (NIHR203312). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. The University of Cambridge has received salary support (JW, ERM) from the NHS in the East of England through the Clinical Academic Reserve.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception of the work and approved the final version of the manuscript. RA contributed clinical information and edited the manuscript. JW wrote the manuscript. EM edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Molecular Pathology of Human Genetic Disease study is approved by the South Birmingham Research Ethics Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Whitworth, J., Armstrong, R. & Maher, E.R. Wilms tumour resulting from paternal transmission of a TRIM28 pathogenic variant—A first report. Eur J Hum Genet 32, 361–364 (2024). https://doi.org/10.1038/s41431-024-01545-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-024-01545-7
This article is cited by
-
Solving medical mysteries with genomics
European Journal of Human Genetics (2024)
