
Abstract
Many human teratogens are associated with a spectrum of congenital anomalies rather than a single defect, and therefore the identification of congenital anomalies occurring together more frequently than expected may improve the detection of teratogens. Thirty-two EUROCAT congenital anomaly registries covering 6,599,765 births provided 123,566 cases with one or more major congenital anomalies (excluding chromosomal and genetic syndromes) for the birth years 2008–2016. The EUROCAT multiple congenital anomaly algorithm identified 8804 cases with two or more major congenital anomalies in different organ systems, that were not recognized as part of a syndrome or sequence. For each pair of anomalies, the odds of a case having both anomalies relative to having only one anomaly was calculated and the p value was estimated using a two-sided Fisher’s exact test. The Benjamini–Hochberg procedure adjusted p values to control the false discovery rate and pairs of anomalies with adjusted p values < 0.05 were identified. A total of 1386 combinations of two anomalies were analyzed. Out of the 31 statistically significant positive associations identified, 20 were found to be known associations or sequences already described in the literature and 11 were considered “potential new associations” by the EUROCAT Coding and Classification Committee. After a review of the literature and a detailed examination of the individual cases with the anomaly pairs, six pairs remained classified as new associations. In summary, systematically searching for congenital anomalies occurring together more frequently than expected using the EUROCAT database is worthwhile and has identified six new associations that merit further investigation.
Similar content being viewed by others


Introduction
Identifying new teratogens is one of the main goals of a congenital anomaly registry. As many known human teratogens are associated with a spectrum of congenital anomalies rather than an isolated anomaly [1,2,3,4,5], identification of cases with multiple congenital anomalies that occur together more frequently than would be expected due to chance alone is likely to be more sensitive in the detection of teratogens. Around three out of four fetuses with a congenital anomaly have an isolated anomaly [6]. Among the remaining fetuses many of these anomalies are due to a chromosomal anomaly (for example, Down syndrome), a single gene defect (for example, Noonan syndrome) or a known teratogen (for example, cytomegalovirus infection), but some fetuses have more than one major anomaly without known etiology. Multiple congenital anomalies may also occur as a consequence of a single primary anomaly (for example, the Potter sequence resulting from renal agenesis and with secondary lung hypoplasia and clubfoot). It is therefore important to identify cases with two or more congenital anomalies in different organ systems, where the pattern of anomalies has not been recognized as part of a syndrome, known association or sequence as these could indicate unknown teratogens or new associations. Around 2% of all births have a congenital anomaly, but multiple congenital anomalies occur in around 16 per 10,000 births, with specific combinations of anomalies being even rarer [7]. Therefore, it is necessary to analyze the data from large datasets with sufficient cases.
The aim of this study was to analyze data from the EUROCAT network of congenital anomaly registries collected from births between 2008 and 2016. A method to automatically identify all pairs or triplets of anomalies occurring more frequently than would be expected due to chance alone was developed. Once such pairs/triplets were identified, the literature was searched to determine if such pairs/triplets of anomalies had already been identified as being part of an association or sequence. Any new pairs of anomalies were examined in greater detail by the registries to determine if any genetic test results were available.
Subjects and methods
The first step in the identification of multiple anomalies is correct case classification. The EUROCAT multiple congenital anomaly algorithm has been developed in collaboration between the EUROCAT Central Registry and the Coding and Classification Committee and continuously improved since 2004 [6]. The members of the Coding and Classification Committee are geneticists and pediatricians. The algorithm classifies cases into different groups based on ICD-10/British Paediatric Association (BPA) codes. The aim of the algorithm is to classify congenital anomaly cases into:
-
(a)
Chromosomal syndromes: all cases where an unbalanced chromosomal anomaly has been diagnosed, irrespective of types of anatomically defined anomalies.
-
(b)
Genetic and environmental syndromes: all cases are due to a single gene defect or a known environmental teratogen, irrespective of types of anatomically defined anomalies. This includes skeletal dysplasias and hereditary skin disorders.
-
(c)
Isolated anomalies: all cases with one congenital anomaly/anomalies occurring in only one organ subgroup or with a known sequence where multiple congenital anomalies cascade as a consequence of a single primary anomaly.
-
(d)
Multiple congenital anomalies: cases with two or more major congenital anomalies in different organ systems, where the pattern of anomalies has not previously been recognized as part of a syndrome or sequence.
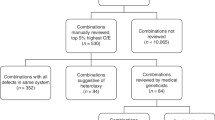
Papers published in 2011 and 2014 describe the methodology and results of the first 2 years of data [6, 7]. The computer algorithm allocates 90% of all EUROCAT cases into classification groups (a), (b) or (c). Approximately 10% of cases are classified by the computer as potential multiple cases and these cases were reviewed by three EUROCAT geneticists to reach an agreement for classification as true multiple congenital anomaly cases (d) or allocation to another group. A web-based system for review of cases has been developed, which allows easy and fast review of many cases and transfer of the final decision back to the central database. If two geneticists agreed on a case classification, this was considered the final decision. If all three geneticists disagreed or one of them classified the case for query, the moderator made the final decision.
Thirty-two full-member registries covering 6,599,765 births provided 154,154 cases with one or more major congenital anomalies born between 2008 and 2016. Cases with chromosomal and genetic syndromes, skeletal dysplasias or hereditary skin disorders were excluded resulting in 123,566 cases for inclusion in this analysis.
Statistical methods
Sixty EUROCAT congenital anomaly subgroups were used in the analysis (Appendix Table A); 57 specific congenital anomaly subgroups and three more general congenital anomaly subgroups (neural tube defects (NTDs), congenital heart defects (CHD) and Severe CHD [8]).
Analysis of multiple congenital anomaly cases only
All cases classified above as multiple congenital anomaly cases were analyzed as follows. For each pair of anomalies (say A and B) the odds of a case having anomaly B given that it had anomaly A relative to the odds of a case having anomaly B given that it did not have anomaly A was calculated and the associated p value estimated using a two-sided Fisher’s exact test. (Note that the odds ratio for anomaly A given anomaly B is identical to the odds ratio for anomaly B given anomaly A—so only one test was performed for each anomaly pair). The relative odds were not calculated for pairs of anomalies included in the same organ or system (for instance ventricular septal defect (VSD) and any other cardiac anomaly). They were also not calculated for clubfoot with spina bifida or renal dysplasia as clubfoot is considered to arise as a result of the occurrence of spina bifida or renal dysplasia. Finally, they were not calculated for situs inversus and any cardiac anomaly as this association is part of the heterotaxy spectrum.
Multiple testing procedures were carried out using the Benjamini–Hochberg procedure to control the false discovery rate. This gave a corrected overall p value to determine statistical significance and thus adjusted p values were calculated. Pairs of anomalies with adjusted p values < 0.05 were examined further. The analysis was repeated for males and females separately as hypospadias is only present in males (33 cases of indeterminate sex and 489 cases with missing sex were excluded).
Logistic regression models were used to examine associations between three anomalies. Each anomaly in turn was regressed on two other anomalies and the interaction term provided an estimate of the odds ratio for all three anomalies given any of the other two anomalies. As before, sets of anomalies known to be related were excluded, and the Benjamini–Hochberg procedure to control the false discovery rate was applied to obtain adjusted p values.
Analysis of all cases with an anomaly
The above analysis was repeated on the population of all anomaly cases (n = 123,566), not just those with multiple anomalies. The number of cases with both anomalies remains the same, but the number of cases with each individual anomaly increases due to the inclusion of cases with only one anomaly. The estimated relative odds were therefore inflated, and the p values reduced. We therefore only examined pairs of anomalies with adjusted p values < 0.01, rather than the <0.05 cut-off used above.
Results of the statistical analysis
The EUROCAT multiple congenital anomaly algorithm followed by a review by three EUROCAT geneticists identified 8804 (7.1%; 95% confidence interval (CI): 7.0–7.3) cases for the 9 years with two or more anomalies out of all 123,566 cases without a genetic disorder. The proportion of multiple anomaly cases was greater in males (7.1%; 95% CI: 6.9–7.3) than in females (6.8%; 95% CI 6.5–7.0), though not statistically significant.
A total of 31 statistically significant positive associations between two EUROCAT subgroups were found when analyzing the cases with multiple congenital anomalies only and judging significance from an adjusted p value < 0.05. These results are very similar to those obtained by analyzing all cases with a congenital anomaly (rather than just those with multiple anomalies) and selecting those associations with an adjusted p value of <0.01 (Tables 1 and 2). The results were also similar when males and females were analyzed separately.
There were no combinations of three anomaly subgroups that were statistically significantly more likely to occur than any of the combinations of two anomalies.
Results of the review by the EUROCAT coding and classification committee
The list of the 31 significant associations was reviewed by the EUROCAT Coding and Classification Committee to determine if the pairs identified were potential unrecognized associations or were part of a known association or sequence or had occurred due to other reasons. Nineteen associations were known associations or sequences already described in the literature (Table 1). Associations were thought to be part of limb body wall complex, OEIS complex (omphalocele-exstrophy-imperforate anus-spinal defects), VACTERL association or sequences like Prune Belly Sequence. One association was explained by coding errors of the anomalies included in the association and related to a known association (also Table 1).
Potential new associations
Eleven associations were determined “unknown” and were selected for literature reviews and reviews of the individual cases with the association by the Coding and Classification Committee members in collaboration with the local registries (Table 2). For some associations such as atrioventricular septal defect and duodenal atresia the registries checked for the most recent karyotype testing that may have been performed after the case was notified to the registry. Details of these investigations including the proportion of cases with known karyotypes are given in Table 2 ordered according to the odds ratios.
Two anomaly pairs, microcephaly and congenital cataract; Ebstein’s anomaly and cleft lip, were judged not to be a new association because most of these cases were suspected of having an undiagnosed genetic disorder. Three anomaly pairs were judged to have weak evidence in the literature of a known association: anencephalus and gastroschisis; NTDs and gastroschisis; encephalocele and cleft lip [9,10,11,12,13]. Six anomaly pairs were judged to have evidence of a new association and will be investigated in further detail during annual surveillance in EUROCAT: encephalocele and anophthalmos/micropthalmos; cleft lip and anophthalmos/microphthalmos; hydrocephaly and hypoplastic right heart; atrioventricular septal defect and duodenal atresia/stenosis; tetralogy of Fallot and duodenal atresia/stenosis; severe CHD and duodenal atresia/stenosis.
Discussion
This study examined 1386 different combinations of two anomalies occurring in the same case and identified 31 significant associations of which 20 were known associations. The remaining 11 significant associations were evaluated in detail and six pairs of anomalies were considered to be new associations and not part of any known association or sequence. The EUROCAT surveillance on these six anomaly pairs will be continued as part of the routine surveillance for clusters and trends.
The classification of significant associations as known associations or sequences was based on published literature. Ten anomaly pairs were part of the limb body wall complex [14, 15], OEIS complex [16, 17] or VACTERL association [18, 19]. The association of neural tube defects and omphalocele was documented previously and explains three of the anomaly pairs found in this study [9, 20]. Newborn infants with diaphragmatic hernia often have pulmonary hypertension [21] which keeps the PDA open explaining one anomaly pair. We classified the anomaly pair posterior urethral valves and clubfoot as the oligohydramnios sequence [22]. The remaining three pairs classified in this study as known associations are less well known but are documented in the literature [23,24,25].
Of the six new associations, three pairs overlap: severe CHD, Tetralogy of Fallot and common AV canal were found to be associated to duodenal atresia. We only found one publication describing a non-genetic association in two siblings [26]. As this association is very well-described for children with Down syndrome, we will follow up on future cases with a special focus on genetic tests performed. However, it is unlikely that Down syndrome will remain undiagnosed in liveborn infants. The new association between encephalocele and an/microphthalmos was rarely found in the literature [27]. The same holds true for an/microphthalmos and cleft lip, which was not found in the literature. The last new pair of anomalies, hydrocephaly and hypoplastic right heart syndrome, only occurred in males and many cases had associated renal/genital anomalies. We found three publications with these two anomalies together of which two described cases with a genetic background [28, 29] and one suggesting this combination of anomalies could be part of VACTERL [30]. Further surveillance will be done for all new associations.
Other studies have used similar approaches to identify new associations. For example, the Co-occurring defect analysis approach recommended by Benjamin et al. [31] and used by Howley et al. [32], is based on a modified observed-to-expected (O/E) ratio of co-occurring birth defects (congenital anomalies) that was originally proposed by Khoury et al. [33]. The method adjusts for the tendency of birth defects to cluster with other major malformations. The data analyzed in our study firstly only compared the occurrence of a pair of anomalies within cases that had at least two anomalies (whereas the studies above included isolated anomalies) and therefore the tendency to cluster did not need to be adjusted for in the first set of analyses. The second analysis compared pairs of anomalies to cases with only one anomaly and as expected the odds ratios were higher. However, when adjusted p values were calculated, a similar set of anomalies was statistically significant at p < 0.01. A second important difference between the method adopted by Benjamin et al. [31] was that in this analysis we excluded any cases with known chromosome or genetic anomalies. We wanted to identify any new anomaly clusters—we were not interested in identifying known associations.
The strength of this study was that it was based on data from 32 EUROCAT full-member congenital anomaly registries covering 6,599,765 births between 2008 and 2016. EUROCAT has standardized methods of coding and data cleaning which are adopted by all member registries and the data quality is monitored by the use of data quality indicators. The EUROCAT multiple congenital anomaly algorithm identifies all cases with two or more non-genetic major congenital anomalies in different organ systems, where the pattern of anomalies has not been recognized as part of an association or sequence. A limitation of the study was that it was not possible to obtain detailed genetic information on all cases—the researchers were dependent on the data that had been provided to the registry as the individual cases could not be contacted for more information.
In summary, most associations found by the statistical analysis were known associations already described in the literature. However, there were six new associations that need continued investigation and will be followed by the annual EUROCAT surveillance system.
Data availability
The data analyzed during this study belong to the individual EUROCAT congenital anomaly registries. Applications to analyze these data will be considered by the JRC-EUROCAT management committee and if they are considered of high scientific merit, permissions for the sharing of the data will be sought by the JRC from the relevant registries.
Code availability
The R data analysis programs are available on reasonable request from JM, the corresponding author.
References
Khoury MJ, Adams MM, Rhodes P, Erickson JD. Monitoring for multiple malformations in the detection of epidemics of birth defects. Teratology. 1987;36:345–53.
Khoury MJ, Botto L, Mastroiacovo P, Skjaerven R, Castilla E, Erickson JD. Monitoring for multiple congenital anomalies: an international perspective. Epidemiol Rev. 1994;16:335–50.
Khoury MJ, Botto L, Waters GD, Mastroiacovo P, Castilla E, Erickson JD. Monitoring for new multiple congenital anomalies in the search for human teratogens. Am J Med Genet. 1993;46:460–6.
Kallen KB, Castilla EE, da Graca Dutra M, Mastroiacovo P, Robert E, Kallen BA. A modified method for the epidemiological analysis of registry data on infants with multiple malformations. Int J Epidemiol. 1999;28:701–10.
Kalter H. Congenital malformations: an inquiry into classification and nomenclature. J Med Genet. 1998;35:661–5.
Garne E, Dolk H, Loane M, Wellesley D, Barisic I, Calzolari E, et al. Paper 5: surveillance of multiple congenital anomalies: implementation of a computer algorithm in European registers for classification of cases. Birth Defects Res Part A Clin Mol Teratol. 2011;91:S44–50.
Calzolari E, Barisic I, Loane M, Morris J, Wellesley D, Dolk H, et al. Epidemiology of multiple congenital anomalies in europe: a EUROCAT population-based registry study. Birth Defects Res Part A Clin Mol Teratol. 2014;100:270–6.
EUROCAT_Central_Registry. Special Report: Congenital Heart Defects in Europe 2000–2005. https://eu-rd-platform.jrc.ec.europa.eu/sites/default/files/eurocat-pub-docs/Special-Report-CHD.pdf2009.
Calzolari E. Omphalocele and gastroschisis in Europe: a survey of 3 million births 1980–1990. EUROCAT working group. Am J Med Genet. 1995;58:7.
Forrester MB, Merz RD. Structural birth defects associated with omphalocele and gastroschisis, Hawaii, 1986–2001. Congenit Anom. 2008;48:87–91.
Ganapathy A, T S, Swer MH, Rao S. Occipital meningoencephalocele with cleft lip, cleft palate and limb abnormalities- a case report. J Clin Diagn Res. 2014;8:Ad03–5.
König M, Due-Tønnessen B, Osnes T, Haugstvedt JR, Meling TR. Median facial cleft with a frontoethmoidal encephalocele treated with craniofacial bipartition and free radial forearm flap: a case report. Skull Base. 2010;20:119–23.
Richieri-Costa A, Zechi-Ceide RM, Candido-Souza RM, Monteiro RAC, Tonello C, de Freitas ML, et al. Holoprosencephaly, orofacial cleft, and frontonaso-orbital encephaloceles: genetic evaluation of a possible new syndrome. Am J Med Genet A. 2019;179:2170–7.
Bergman JEH, Barišić I, Addor M-C, Braz P, Cavero-Carbonell C, Draper ES, et al. Amniotic band syndrome and limb body wall complex in Europe 1980–2019. Am J Med Genet Part A. 2023;191:995–1006.
Bugge M. Body stalk anomaly in Denmark during 20 years (1970–1989). Am J Med Genet Part A. 2012;158A:1702–8.
Arteaga-Vázquez J, Luna-Muñoz L, Morales-Suárez JJ, Mutchinick OM. OEIS complex: prevalence, clinical, and epidemiologic findings in a multicenter Mexican birth defects surveillance program. Birth Defects Res. 2019;111:666–71.
Keppler-Noreuil K M. OEIS complex (omphalocele-exstrophy-imperforate anus-spinal defects): a review of 14 cases. Am J Med Genet. 2001;4:217–9.
Botto LD, Khoury MJ, Mastroiacovo P, Castilla EE, Moore CA, Skjaerven R, et al. The spectrum of congenital anomalies of the VATER association: an international study. Am J Med Genet. 1997;71:8–15.
van de Putte R, van Rooij IALM, Marcelis CLM, Guo M, Brunner HG, Addor M-C, et al. Spectrum of congenital anomalies among VACTERL cases: a EUROCAT population-based study. Pediatr Res. 2020;87:541–9.
Calzolari E, Bianchi F, Dolk H, Stone D, Milan M. Are omphalocele and neural tube defects related congenital anomalies?: data from 21 registries in Europe (EUROCAT). Am J Med Genet. 1997;72:79–84.
Mohseni-Bod H, Bohn D. Pulmonary hypertension in congenital diaphragmatic hernia. Semin Pediatr Surg. 2007;16:126–33.
Christianson C, Huff D, McPherson E. Limb deformations in oligohydramnios sequence: effects of gestational age and duration of oligohydramnios. Am J Med Genet. 1999;86:430–3.
Husmann DA. Lessons learned from the management of adults who have undergone augmentation for spina bifida and bladder exstrophy: incidence and management of the non-lethal complications of bladder augmentation. Int J Urol. 2018;25:94–101.
Moosa S, Lambie LA, Krause A. Sirenomelia: four further cases with discussion of associated upper limb defects. Clin Dysmorphol. 2012;21:124–30.
Ohuchi H, Sato K, Habuta M, Fujita H, Bando T. Congenital eye anomalies: more mosaic than thought? Congenit Anom. 2019;59:56–73.
Lemire EG, Evans JA, Giddins NG, Harman CR, Wiseman NE, Chudley AE. A familial disorder with duodenal atresia and tetralogy of Fallot. Am J Med Genet. 1996;66:39–44.
Boahene DKO, Bartley GB, Clay RP, Thompson DM. Heminasal proboscis with associated microphthalmos and encephalocele. J Craniofac Surg. 2005;16:300–6.
Henn W, Reichert H, Nienhaus H, Zankl M, Lindinger A, Hoffmann W, et al. Progressive hydrocephalus in two members of a family with autosomal dominant Noonan phenotype. Clin Dysmorphol. 1997;6:153–6.
Leonardi ML, Pai GS, Wilkes B, Lebel RR. Ritscher-Schinzel cranio-cerebello-cardiac (3C) syndrome: report of four new cases and review. Am J Med Genet. 2001;102:237–42.
Chung B, Shaffer LG, Keating S, Johnson J, Casey B, Chitayat D. From VACTERL-H to heterotaxy: variable expressivity of ZIC3-related disorders. Am J Med Genet A. 2011;155a:1123–8.
Benjamin RH, Yu X, NavarroSanchez ML, Chen H, Mitchell LE, Langlois PH. et al. Co-occurring defect analysis: a platform for analyzing birth defect co-occurrence in registries. Birth Defects Res. 2019;18:1356–64.
Howley MM, Williford E, Agopian AJ, Lin AE, Botto LD, Cunniff CM, et al. Patterns of multiple congenital anomalies in the National Birth Defect Prevention Study: challenges and insights. Birth Defects Res. 2023;115:43–55.
Khoury MJ, James LM, Erickson JD. On the measurement and interpretation of birth defect associations in epidemiologic studies. Am J Med Genet. 1990;37:229–36.
Acknowledgements
We would like to acknowledge the work of all the people involved in the EUROCAT Congenital Anomaly Registries for ensuring the availability of such high-quality surveillance data. In addition, we would like to acknowledge the work of James Densem in creating software to enable the examination of individual cases with multiple anomalies.
Funding
No financial assistance was received in support of the study. Each EUROCAT registry is responsible for its own funding for surveillance.
Author information
Authors and Affiliations
Contributions
JEHB, IB, DW, DT, EG: members of the EUROCAT Coding and Classification Committee and all have contributed to the creation of the algorithm for identifying which are multiple Cas, have been involved in determining the final classification of the congenital anomaly cases and have performed literature reviews investigating the new multiples identified. They all provided feedback on the draft manuscript. JKM: performed the statistical analysis and wrote the first draft and all subsequent drafts. EL: performed the statistical analysis and provided feedback on the draft manuscript. M-CA, CC-C, CMD, ESD, LJE-G-d-g, MG, KK, NL, KL, AM-K, VN, AN, IP, AP, HR-R, JR, AR, FR, GS, WW, AK-O: provided the data and feedback on the draft manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morris, J.K., Bergman, J.E.H., Barisic, I. et al. Surveillance of multiple congenital anomalies; searching for new associations. Eur J Hum Genet 32, 407–412 (2024). https://doi.org/10.1038/s41431-023-01502-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01502-w
This article is cited by
-
Artificial intelligence â the next generation of sequencing?
European Journal of Human Genetics (2024)
