
Abstract
The neuronal SNARE complex drives synaptic vesicle exocytosis. Therefore, one of its core proteins syntaxin 1A (STX1A) has long been suspected to play a role in neurodevelopmental disorders. We assembled eight individuals harboring ultra rare variants in STX1A who present with a spectrum of intellectual disability, autism and epilepsy. Causative variants comprise a homozygous splice variant, three de novo missense variants and two inframe deletions of a single amino acid. We observed a phenotype mainly driven by epilepsy in the individuals with missense variants in contrast to intellectual disability and autistic behavior in individuals with single amino acid deletions and the splicing variant. In silico modeling of missense variants and single amino acid deletions show different impaired protein-protein interactions. We hypothesize the two phenotypic courses of affected individuals to be dependent on two different pathogenic mechanisms: (1) a weakened inhibitory STX1A-STXBP1 interaction due to missense variants results in an STX1A-related developmental epileptic encephalopathy and (2) a hampered SNARE complex formation due to inframe deletions causes an STX1A-related intellectual disability and autism phenotype. Our description of a STX1A-related neurodevelopmental disorder with or without epilepsy thus expands the group of rare diseases called SNAREopathies.
Similar content being viewed by others
Introduction
SNARE (soluble NSF attachment protein receptor) complexes play a crucial role in a multitude of membrane fusion processes in the central nervous system. The core complex, comprising neuronal SNAREs syntaxin 1, SNAP25 and VAMP2, mediates synaptic vesicle exocytosis [1] as well as secretion of neuropeptides and neurotrophins [2]. Syntaxin 1A (encoded by the gene STX1A) is localized at the neuronal synaptic plasma membrane. It is involved in regulation of several plasma membrane-bound monoamine transporters [3,4,5,6]. Variants in STX1A have not been shown to cause a Mendelian disorder. Of note, de novo deletions of STX1A was seen in 5 out of 83 individuals with autism spectrum disorder (ASD) [7] although a recent analysis of a large ASD cohort did not show a significant association [8]. Single nucleotide polymorphisms in STX1A are suspected to increase susceptibility to migraine in several case-control-studies [9,10,11,12] as well as playing a role in predisposition to autism [13,14,15], cryptogenic epilepsy [16] and children attention-deficit/hyperactivity disorder [17], whereas no associations could be drawn to schizophrenia [18].
Due to little redundancy and rescue mechanisms, disruption of components of the SNARE complex give rise to a group of rare diseases called SNAREopathies [19, 20]. In this study, we describe eight individuals harboring ultra rare homozygous or de novo heterozygous variants in STX1A to delineate a novel STX1A-related neurodevelopmental disorder based on different pathogenic mechanisms and inheritance models.
Materials (Subjects) and methods
Standard protocol approvals
The study was approved by the ethics committee of the University of Leipzig, Germany (402/16-ek). All families provided informed consent for clinical phenotyping, genetic testing and publication. Testing and research analysis was approved by local ethics committees in the respective institutions.
Research cohort and identification of variants
Eight individuals with STX1A variants from seven different families were assembled via literature review and GeneMatcher [21]. Phenotypic and genotypic information was obtained from the referring collaborators using a standardized questionnaire, evaluating family anamnesis, clinical history, genetic testing, variant details, EEG, brain imaging and medication (see STX1A_Supplementary Table S1). All individuals underwent exome or genome sequencing. Since no causative variants in a known disease gene were identified [22], the data were examined in a scientific approach, including parental sequence data if available. Maternity and paternity was proved for all de novo variants. All variants were prioritized considering allele frequency in gnomAD [23] below 1%, impact on protein function via different in silico programs (Supplementary Tables S2 and S3) and involvement of candidate genes in neuronal processes. Apart from the STX1A variants observed, none of the individuals described had other remarkable findings that likely explain the phenotype. All variants in STX1A are described with regard to GRCh37 (NM_004603.4) and have been classified according to the ACMG criteria (Supplementary Table S4) [24].
Structural modeling
Structural analysis of the variants was performed based on the crystal structure of Syntaxin 1A in complex with Syntaxin-binding protein 1 (PDB: 3C98) [25] or the components of the SNARE complex (PDB: 6IP1, 6MDN) [26, 27]. Amino acid changes were modeled with SwissModel [28] and RasMol [29] was used for structure analysis and visualization.
Results
We describe eight individuals harboring ultra rare variants in STX1A. All variants were absent from the gnomAD database [23].
Clinical description
The eight individuals exhibited varying neurodevelopmental, neurological and neuropsychiatric symptoms. In four out of eight individuals, epilepsy was the leading clinical symptom, whereas in the four remaining individuals intellectual disability and autistic behavior without epilepsy was prominent. An overview of the clinical symptoms is presented in Table 1 and Supplementary Table S1.
Individual 1: This male was first reported in Reuter et al. [30]. He was born as first of ten children to healthy consanguineous Syrian parents (cousins of 1° degree) without intellectual disability, behavior abnormalities or seizures in medical history. One of his brothers (described below as individual 2) presented with similar symptoms. Another brother had died in infancy due to epidermolysis bullosa-like symptoms (see Supplementary Fig. S1). During pregnancy of individual 1 decreased fetal movements were noted. He was born at term with increased birth length and neonatal hypotonia. He learned unassisted walking at the age of three years and did not show persistent motor problems in later childhood. Global development and speech were delayed. Clinical assessment at the age of 24 years showed moderate intellectual disability with mostly unremarkable speech and behavior. Seizures were denied. MRI or EEG were not performed. No facial dysmorphisms were seen. He was able to fulfill easy tasks on the family’s agricultural holding.
Individual 2: This male was also first reported in Reuter et al. [30] and is the younger brother of individual 1. He was born as the third child in the family described above. As in his brother, decreased fetal movements were noted in pregnancy. After birth at term, neonatal hypotonia and increased birth length were noted. He also learned unassisted walking around age three years and did not show persistent motor problems in later childhood. Global development and speech were delayed. Clinical assessment at the age of 20 years showed moderate intellectual disability, but a mostly unremarkable speech. His behavior was more aggressive compared to his brother. No seizures were recorded. Neither MRI nor EEG were performed. No facial dysmorphisms were seen. As his brother he was able to fulfill easy tasks on the family’s agricultural holding.
Exome sequencing detected a homozygous variant in STX1A:c.284-1G>A in both brothers. The parents and the second oldest sister were found to be heterozygous for this variant via Sanger sequencing. The remaining unaffected siblings were not tested for carrier status. Unfortunately, this family was lost to further follow-ups.
Individual 3: This male was born after an unremarkable pregnancy to healthy parents. Birth measurements were around the 50th percentile and neonatal hypotonia was observed. This individual was affected by severe global developmental delay. He learned unassisted walking at the age of three years and six months, but had persistent motor problems in later childhood. Clinically, he was suspected to harbor an Angelman-like disorder. He was able to speak two to three words at the age of three, but speech was absent at last clinical assessment at the age of 14 years. His intellectual disability was assessed to be severe. Abnormal behavior comprised stereotypes, drooling, bruxism, biting and pulling hair and he was diagnosed with ASD. He also had recurrent sleeping problems. Neurological examination showed ptosis, hypotonia, ataxia and spasticity with pyramidal signs. First tonic-clonic seizures were noticed at the age of six months, reaching a frequency of several per day. A therapy with valproate and levetiracetam was successful to suppress severe seizures for several years, although absences and rare tonic seizures remained. At the age of 12 years, seizures intensified again, for unknown reasons, reaching frequencies of several tonic and tonic-clonic seizures and absences per day. A switch in medication to rufinamide, valproate and perampanel lead to a limited response with suppressing seizure frequency to one per week. MRIs were normal apart from delayed myelination at three years of age. Family history only elicited a febrile seizure in his maternal uncle at the age of 1 year.
Trio exome sequencing detected a heterozygous de novo variant in STX1A:c.435C>G, p.(Cys145Trp).
Individual 4: A now 9 year old girl, was born after an uneventful pregnancy at 39 weeks of gestation. Birth weight was on the 75th percentile. First seizures occurred on day 1 of life day, which was attributed to neonatal hypoglycemia (1.4 mmol/L glucose) due to diazoxide responsive congenital hyperinsulinism. She spent 5 weeks in the neonatal intensive care unit. After the neonatal period, she was seizure-free until 10 months of age, when she presented with febrile seizures, and shortly after recurrent afebrile focal seizures (2–3 per week). Subsequently she developed focal to bilateral tonic-clonic, absence and generalized onset motor tonic-clonic seizures. At age of seven years, she presented with myoclonic, myoclonic absence and focal onset (occipital lobes) seizures. Pharmacological treatment included valproate, phenobarbitone, pyridoxine, levetiracetam, carbamazepine, lamotrigine, zonisamide, clobazam, lacosamide, rufinamide, ethosuximide. Her current seizure frequency is approximately 2 absences and 1 focal seizure per month on sodium valproate and ethosuximide. At the age of nine years, weight was on the 50th to 75th percentile. Concerning her cognitive development, she started babbling aged 8 months but then stopped at 9 months, thus indicating a regression of skills. She has severe ID and never acquired speech, but can use picture books to communicate. She walked independently aged 3 ½ years, but has persistent motor problems in later childhood with a waddling gait. She has autistic traits but does not meet the criteria for an ASD diagnosis and she was diagnosed with sensory processing disorder. The latest EEG at five years of age showed a slow background, multifocal epileptiform spikes, and occipital spike waves and generalized irregular-semi-irregular spike waves. MRI performed at four years of age showed mild periventricular leukoencephalopathy. In family history, the mother had two seizures as a teenager and was treated with Carbamazepine for two years, after which she was weaned. No further seizures occurred.
Trio genome sequencing detected a heterozygous de novo variant in STX1A: c.435C>G, p.(Cys145Trp).
Individual 5: This male was born at term after an unremarkable pregnancy. His birth length was 49 cm (14th percentile), weight 2800 gram (12th percentile) and head circumference 34 cm (17th percentile). Neonatal hypotonia was noticed. He had global developmental delay and persistent motor problems throughout his life, but learnt unassisted walking at the age of 14 months. His first speech was noted at the age of 36 months. At the age of nine years and six months, his speech was mildly age-inappropriate, reaching 5th centile (Bayley Scales of Infant Development). Moderate intellectual disability as well as behavioral problems such as aggressiveness, hyperactivity, attention deficit and sleeping problems were reported. He had first seizures (loss of consciousness) at the age of seven years and four months with a frequency of one seizure every two to three months. He exhibited right frontal to centero-temporal spikes in EEG and was diagnosed with rolandic epilepsy. Antiepileptic therapy was started at the age of 8 years and consisted of Levetiracetam. During therapy, no further seizures occurred, but the EEG anomalies persisted. Neurological examination showed fine intentional tremors as well as global motor impediment. A brain MRI at the age of five years had normal results. Furthermore, axial hypotonia, a congenital clubfoot and various mild dysmorphisms, including brachycephaly, low set ears, retrognathia, tapered fingers, lower limb muscular hypotrophy and hypotenar eminence as well as hypochromic and coffee-milk skin spots were noted. His weight reached 97th percentile, OFC was at the 36th percentile. In family history, the mother was diagnosed with migraine and a Chiari malformation.
Trio exome sequencing detected a heterozygous de novo variant in STX1A: c.554C>G, p.(Ser185Cys).
Individual 6: This patient had moderate intellectual disability. No seizures were recorded. Unfortunately, this patient was lost to follow-up for further detailed phenotyping. Exome sequencing detected a heterozygous variant in STX1A: c.668_670del. p.(Val223del) of unknown origin due to unavailable parental testing.
Individual 7: This female was born to healthy parents after 42 weeks of an unremarkable pregnancy. Birth weight was 3300 g, neonatal hypotonia was noted. At the age of 6 months of age, body measurements were: 6085 g (11th percentile), 66 cm (46th percentile) and OFC 41 cm (9th percentile). At last follow-up at the age of 14 years measurements were: 38 kg (2nd percentile), 150 cm (3rd percentile). She had severe global developmental delay, motor problems (she never learned walking), absence of speech and profound intellectual disability. Neurological examination showed dysphagia, drooling and spastic paresis. Epileptic spasms became evident at the age of three months and were associated with hypsarrhythmia on EEG, compatible with West syndrome. Additionally, daily tonic seizures occurred and a severe epileptic encephalopathy developed. Treatment included ACTH, valproate, levetiracetam, clonazepam and cannabidiol, suppressing frequency to weekly intervals but without reaching longer seizure free periods. The encephalopathic phenotype later progressed to Lennox-Gastaut syndrome. A brain MRI at the age of three years showed normal results.
Trio exome sequencing detected a heterozygous de novo variant in STX1A: c.677A>G, p.(Gln226Arg).
Individual 8: This male was born at term to healthy parents after an unremarkable pregnancy. Birth measurements determined a length of 50 cm (8th percentile) and a weight of 3000 g (14th percentile). Neonatal hypotonia was observed. At age of 24 months he showed a length of 71 cm (<1st percentile) and a weight of 9 kg (1st percentile). At 12 years of age length was at −3 SD while OFC was at +2 SD. This individual was affected by severe intellectual disability with only few words at last assessment and motor delay with unassisted walking at around four years of age. Behavioral problems compatible with an autism spectrum disorder were noted, including an extreme aggressiveness and sleeping problems. Apart from neonatal hypotonia no further neurological abnormalities could be elicited. The individual had no seizures and brain MRI showed normal results. Additional symptoms included a pes varus and constipation.
Trio exome sequencing showed a heterozygous de novo variant in STX1A: c.722_724del, p.(Val241del).
Genotypic spectrum
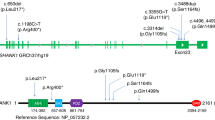
One homozygous splicing variant, three different de novo missense variants and two deletions of a single amino acid (one de novo) were observed in STX1A (Fig. 1). Variants were predicted to be damaging according to multiple in silico tools (Supplementary Tables S2 and S3). The only recurrent variant is the missense variant p.(Cys145Trp) twice occurring de novo in individuals with a consistent phenotype. Furthermore, in the most recent DDD study [31], the de novo variant c.236T>G, p.(Met79Arg) was reported in an individual with a neurodevelopmental disorder, but a detailed phenotypic description is not available.

a Schematic depiction of STX1A. Segments of functional importance are displayed colored and named. The lollipop bars indicate observed causative variants of this study. The de novo variant p.(Met79Arg) was identified in the recent DDD study but a detailed clinical description is lacking for this individual [31]. b Association of observed variants with a tolerance landscape of STX1A created by Metadome [45]. Height and color of the graph indicate tolerance towards variation in the respective amino acid residues: the lower and more red the bar, the less tolerant to variation is the specific residue. c Comparison of selected homologous amino acid residues of STX1A and STX1B. Colored positions mark residues with causative/pathogenic variants in the respective protein.
Structural modeling
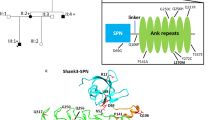
All residues affected by a variant in the present study are resolved in the crystal structure of STX1A in complex with syntaxin-binding protein 1 (STXBP1) [25]. In this complex, residues 29–237 of STX1A form a rather compact structure comprising four helices (Fig. 2A). This conformation was termed ‘closed’ conformation in the literature [25]. The major effect of the missense variants are steric clashes, which destabilize the STX1A structure and are therefore expected to hamper interaction with STXBP1. One example is p.(Cys145Trp), where the bulkier tryptophan causes steric clashes with residues Tyr141 and Arg198. In contrast, such a destabilization of the structure is not expected for the p.(Cys145Ser) exchange that is reported 3x in gnomAD, which does not increase the sidechain volume and consequently leads to no steric clashes.

A Structure of STX1A in complex with STXBP1 (PDB: 3C98). STXBP1 and STX1A are colored in red and cyan/blue, respectively, with the H3 domain of STX1A highlighted in blue. The residues at the sites of mutation are shown in space-filled presentation and colored according to the atom types. B Structure of STX1A as part of the SNARE complex (PDB: 6IP1). STX1A, vesicle-associated membrane protein 2, SNAP25, and α-SNAP are colored in blue, orange, green, and white, respectively. The residues at the sites of mutation are shown in space-filled presentation and colored according to the atom types. Note that only three of the variants investigated are locate in the SNARE-interacting region.
A negative structural effect is also expected for the deletion of p.(Val223del), which disrupts the helical structure of the H3 domain (residues 186–253). In contrast, p.(Val241del) plays only a minor role for binding to STXBP1. An experimental investigation proved that deletion of syntaxin1A residues 241–262 caused only a very moderate decrease in STXBP1 affinity by a factor of two [25].
For a structural interpretation of variant effects, it is important to note that STX1A can also adopt a structurally distinct ‘open’ conformation as part of the SNARE complex. In this complex, the H3 domain forms a long central helix that interacts with the other components of the SNARE complex (Fig. 2B). Both deletion variants p.(Val223del), p.(Val241del) are located in the central part of this helix. One-residue deletions at these sites significantly affect the helix structure and are expected to hamper SNARE complex formation. The effect of the p.(Gln226Arg) variant is known from experiment; site-directed mutagenesis revealed that this exchange results a significantly slower dissociation of the SNARE complex [32].
Response to antiepileptic treatment
All four individuals exhibiting seizures underwent pharmacological treatment. Overall, no active substance was able to suppress seizures for long periods or normalize EEG completely. Therapy response was different between the affected individuals: in individual 3, therapy response to valproate and levetiracetam was good in childhood, but could not prevent absences and rare tonic seizures but had to be altered in puberty to rufinamide, valproate and perampanel, with limited response. In individual 4, valproate, phenobarbitone, pyridoxine, levetiracetam, carbamazepine, lamotrigine, zonisamide, clobazam, lacosamide, rufinamide, ethosuximide were tried as medication, showing a limited response on sodium valproate and ethosuximide. Individual 5 was seizure free under therapy with levetiracetam, although EEG abnormalities remained. In individual 7, ACTH, valproate, levetiracetam, clonazepam and cannabidiol suppressed seizure frequency but without reaching longer seizure free periods.
Discussion
In this study, we delineate a novel STX1A-related neurodevelopmental disorder with or without epilepsy based on two different pathogenic mechanisms and inheritance models.
Given the data from the gnomAD database, STX1A is a gene with a reduced number of missense (z score = 2.4, observed/expected ratio = 0.51 (0.43–0.61)) and truncating/splice variants (pLI score = 0.98 observed/expected ratio = 0.06 (0.02–0.29)). This indicates a selective constraint on those types of variants in a healthy control population that lacks severe early onset phenotypes such as intellectual disability [23]. All variants of this study are absent from the GnomAD database and all except the inherited splicing and the p.(Val223del) variant of unknown origin were de novo. Moreover, the causality of the variants is supported by strong in silico data [33, 34] (Supplementary Tables S2 and S3) and the structural modeling performed in this study. In addition, the phenotypic presentation was consistent in the two patients with the recurring de novo variant and across variant types (missense variants and inframe deletions), albeit in a small cohort. Therefore, different lines of evidence support a causal role of ultra rare variants in STX1A to be causative of a neurodevelopmental disorder with or without epilepsy.
In all individuals with ultra rare variants in STX1A, intellectual disability, neonatal hypotonia and motor delay were present. It appears as if two different phenotypic courses are existing, following the dichotomy of the identified variants and structural modeling: (1) an epileptic encephalopathy in all individuals with missense variants and (2) a neurodevelopmental disorder with primarily intellectual disability and autistic behavior, but no epilepsy in the individuals with an inframe deletion or the family with the homozygous splice variant. The latter resembles the clinical presentation of STX1A haploid individuals in a Japanese ASD cohort [7] where the probands exhibited normal or only mildly impaired intelligence with no epilepsy, but no deep phenotyping was performed. Epilepsy is also rare in individuals with Williams-Beuren-Syndrome [35], who often lack one allele of STX1A, and in whom differences in transcript levels of STX1A have been shown to account for 15.6% of cognitive variation [36, 37]. Only one individual with a high quality heterozygous canonical splice site variant is listed in GnomAD_v2 and v3 respectively (c.466+2T>C, p.?) and therefore unlikely to be affected by intellectual disability. Variants affecting the canonical splice site can result in different effects on pre-mRNA splicing, but usually lead to exon skipping that would result in a frameshift in the case of family 1 and for the variant c.466+2T>C [38]. Thus, it appears likely that monoallelic loss of STX1A leads to varying defects of cognition and social behavior without necessarily causing severe early onset symptoms. Assuming an incomplete splice defect that encompasses the expression of a fraction of normal protein could explain the recessive inheritance mode in family 1 and why severe symptoms became only evident in the two homozygous siblings and not in the heterozygous parents. Epilepsy only occurs in individuals with missense variants in STX1A that are predicted to disturb the interaction of STX1A with STXBP1. Since both loss and gain of function variants in STXBP1 are a known cause of a developmental and epileptic encephalopathy [39], it is tempting to speculate that the phenotypic differences in STX1A are also a result of loss- and gain-of-function effects in protein function. This will have to be examined in follow-up functional studies of the causative variants.
In established SNAREopathies, the most common symptoms comprise neurodevelopmental delay in domains of speech, language, motor function and intellectual ability. Additionally, other neurological symptoms such as seizures, spasms and ataxia as well as social behavioral abnormalities are observed. Severity and frequency of specific symptoms vary depending on the affected gene [19]. The phenotype of the probands in the presented cohort in STX1A is therefore compatible with known SNARE-associated disorders caused by variants in SNAP25, VAMP2, STX1B and STXBP1.
Comparison of STX1A and the closely related STX1B is of special help for variant interpretation. The proteins show identical amino acids at more than 82 percent of positions [40]. Mostly truncating, but also several missense variants in STX1B are known to cause a childhood epilepsy syndrome with both febrile and afebrile seizures, typically with a benign course and good response to treatment. Intelligence is usually normal, although more severely affected individuals have been described [41]. Interestingly, missense variants are described to cause a more severe phenotype than null variants [42]. For two of STX1A missense variants described in the present cohort, pathogenic variation in corresponding STX1B residues are described: In case of the STX1A variant p.(Cys145Trp), a missense variant affecting the homologous residue in STX1B: p.(Cys144Phe) was previously described as likely pathogenic [42]. Interestingly, this individual showed additional features such as tremor and cerebellar ataxia that are not typical symptoms of STX1B-related generalized epilepsy with febrile seizures. For the STX1A variant p.(Gln226Arg) in an individual with West syndrome and Lennox-Gastaut syndrome, the homologous residue directly adjacent is affected in STX1B: p.(Gly226Arg) by a de novo variant in an individual with yet another developmental and epileptic encephalopathy with myoclonic-atonic epilepsy [41].
Attempts to examine clinical relevance via Stx1a ablated mice (null and heterozygous deletion) showed atypical social behavior and abnormal recognition profiles in a dose dependent manner [7, 43, 44]. The observation of autistic symptoms in mice is another line of evidence that nonfunctional STX1A-variants rather cause a phenotype of autism and intellectual disability in humans.
STX1A adopts at least two distinct conformations; a ‘closed’ inhibitory conformation in complex with STXBP1 (Fig. 2A) and an ‘open’ conformation in the SNARE complex (Fig. 2B). In particular, the H3 domain (residues 186–253) forms entirely different interactions in both complexes. When bound to STXBP1, the H3 domain interacts with the N-terminal part of STX1A, rendering it inaccessible to its partner molecules in the SNARE complex. Therefore, incorporation of STX1A into a SNARE complex requires dissociation from STXBP1 and switch to an open conformation, in which the H3 domain is accessible [25]. Consequently, variants may either affect the STX1A-STXBP1 complex and/or formation of the SNARE complex. Based on the structural modeling, all variants except p.(Val241del) cause a destabilization of the STX1A-STXBP1 complex thereby shifting the conformational equilibrium towards the open STX1A conformation. A reduced ability to form a functional SNARE complex is expected for the inframe deletions p.(Val223del) and p.(Val241del). In contrast, the missense variant p.(Gln226Arg) stabilizes this complex by causing a slower dissociation [32]. These different effects on SNARE complex functioning (stabilization vs. destabilization) also offer an explanation for the different phenotype of p.(Gln226Arg) compared to the individuals harboring a deletion.
In the light of the considerations above, the phenotype of intellectual disability with epilepsy is most likely correlated with variants that weaken the inhibitory STX1A-STXBP1 interaction and do not disrupt STX1A function in SNARE complex formation. In contrast, the deletion variants, which mainly hamper SNARE complex formation, result in a different phenotype of intellectual disability and autism without seizures.
Conclusion
In summary, different lines of evidence presented here support that ultra rare heterozygous and homozygous variants in STX1A cause a neurodevelopmental disorder with two different phenotypic presentations: (1) an STX1A-related developmental epileptic encephalopathy due to missense variants weakening the inhibitory STX1A-STXBP1 interaction and (2) an STX1A-related intellectual disability and autism phenotype due to inframe deletions hampering SNARE complex formation. Our description thus expands the group of disorders called SNAREopathies.
Data availability
All data concerning this work is included in the manuscript and its supplement. All variants included in the manuscript have been uploaded to ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679128/. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679133/. https://www.ncbi.nlm.nih.gov/clinvar/variation/984555/. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679129/. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679130/. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679131/. https://www.ncbi.nlm.nih.gov/clinvar/variation/1679132.
References
Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–7.
Arora S, Saarloos I, Kooistra R, van de Bospoort R, Verhage M, Toonen RF. SNAP-25 gene family members differentially support secretory vesicle fusion. J Cell Sci. 2017;130:1877–89.
Sung U, Apparsundaram S, Galli A, Kahlig KM, Savchenko V, Schroeter S, et al. A regulated interaction of Syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J Neurosci. 2003;23:1697–709.
Quick MW. Regulating the conducting states of a mammalian serotonin transporter. Neuron. 2003;40:537–49.
Quick MW. The role of SNARE proteins in trafficking and function of neurotransmitter transporters. In: Sitte HH, Freissmuth M, editors. Neurotransmitter transporters (Handbook of Experimental Pharmacology). Berlin, Heidelberg: Springer; 2006. p. 181–96. https://doi.org/10.1007/3-540-29784-7_9.
Carvelli L, Blakely RD, DeFelice LJ. Dopamine transporter/syntaxin 1A interactions regulate transporter channel activity and dopaminergic synaptic transmission. PNAS. 2008;105:14192–7.
Kofuji T, Hayashi Y, Fujiwara T, Sanada M, Tamaru M, Akagawa K. A part of patients with autism spectrum disorder has haploidy of HPC-1/syntaxin1A gene that possibly causes behavioral disturbance as in experimentally gene ablated mice. Neurosci Lett. 2017;644:5–9.
Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S, et al. Rare coding variation illuminates the allelic architecture, risk genes, cellular expression patterns, and phenotypic context of autism. medRxiv; 2021. http://medrxiv.org/lookup/doi/10.1101/2021.12.20.21267194.
Lemos C, Pereira-Monteiro J, Mendonça D, Ramos EM, Barros J, Sequeiros J, et al. Evidence of Syntaxin 1A involvement in migraine susceptibility: a portuguese study. Arch Neurol. 2010;67. http://archneur.jamanetwork.com/article.aspx?doi=10.1001/archneurol.2010.37.
Kowalska M, Prendecki M, Kapelusiak-Pielok M, Grzelak T, Łagan-Jędrzejczyk U, Wiszniewska M, et al. Analysis of genetic variants in SCN1A, SCN2A, KCNK18, TRPA1 and STX1A as a possible marker of migraine. Curr Genomics. 2020;21:224–36.
Tropeano M, Wöber-Bingöl C, Karwautz A, Wagner G, Vassos E, Campos-de-Sousa S, et al. Association analysis of STX1A gene variants in common forms of migraine. Cephalalgia. 2012;32:203–12.
Corominas R, Ribasés M, Cuenca-León E, Narberhaus B, Serra SA, del Toro M, et al. Contribution of syntaxin 1A to the genetic susceptibility to migraine: a case-control association study in the Spanish population. Neurosci Lett. 2009;455:105–9.
Nakamura K, Anitha A, Yamada K, Tsujii M, Iwayama Y, Hattori E, et al. Genetic and expression analyses reveal elevated expression of syntaxin 1A (STX1A) in high functioning autism. Int J Neuropsychopharmacol. 2008;11:1073–84.
Nakamura K, Iwata Y, Anitha A, Miyachi T, Toyota T, Yamada S, et al. Replication study of Japanese cohorts supports the role of STX1A in autism susceptibility. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:454–8.
Durdiaková J, Warrier V, Banerjee-Basu S, Baron-Cohen S, Chakrabarti B. STX1A and Asperger syndrome: a replication study. Mol Autism. 2014;5:14.
Baghel R, Grover S, Kaur H, Jajodia A, Parween S, Sinha J, et al. Synergistic association of STX1A and VAMP2 with cryptogenic epilepsy in North Indian population. Brain Behav. 2016;6:e00490.
Wang M, Gu X, Huang X, Zhang Q, Chen X, Wu J. STX1A gene variations contribute to the susceptibility of children attention-deficit/hyperactivity disorder: a case–control association study. Eur Arch Psychiatry Clin Neurosci. 2019;269:689–99.
Kawashima K, Kishi T, Ikeda M, Kitajima T, Yamanouchi Y, Kinoshita Y, et al. No association between tagging SNPs of SNARE complex genes (STX1A, VAMP2 and SNAP25) and schizophrenia in a Japanese population. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1327–31.
Verhage M, Sørensen JB. SNAREopathies: diversity in mechanisms and symptoms. Neuron. 2020;107:22–37.
Klöckner C, Sticht H, Zacher P, Popp B, Babcock HE, Bakker DP, et al. Correction to: de novo variants in SNAP25 cause an early-onset developmental and epileptic encephalopathy. Genet Med. 2021;23:796.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Jauss R-T, Popp B, Platzer K, Jamra R. MorbidGenes-Panel-v2022-02.1. Zenodo; 2022. https://zenodo.org/record/6136995.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 2008;27:923–33.
Huang X, Sun S, Wang X, Fan F, Zhou Q, Lu S, et al. Mechanistic insights into the SNARE complex disassembly. Sci Adv. 2019;5:eaau8164.
White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT. Structural principles of SNARE complex recognition by the AAA+ protein NSF. Elife. 2018;7:e38888.
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23.
Sayle RA, Milner-White EJ. RASMOL: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374.
Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C, et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry. 2017;74:293–9.
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62.
Scales SJ, Yoo BY, Scheller RH. The ionic layer is required for efficient dissociation of the SNARE complex by α-SNAP and NSF. PNAS. 2001;98:14262–7.
Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13:31.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Nicita F, Garone G, Spalice A, Savasta S, Striano P, Pantaleoni C, et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am J Med Genet A. 2016;170A:148–55.
Gao MC, Bellugi U, Dai L, Mills DL, Sobel EM, Lange K, et al. Intelligence in Williams Syndrome is related to STX1A, which encodes a component of the presynaptic SNARE complex. PLoS One. 2010;5:e10292.
Botta A, Novelli G, Mari A, Novelli A, Sabani M, Korenberg J, et al. Detection of an atypical 7q11.23 deletion in Williams syndrome patients which does not include the STX1A and FZD3 genes. J Med Genet. 1999;36:478–80.
Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59:253–68.
Lammertse HCA, van Berkel AA, Iacomino M, Toonen RF, Striano P, Gambardella A, et al. Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain. 2020;143:441–51.
Align results. 2021, https://www.uniprot.org/align/A202108214ABAA9BC7178C81CEBC9459510EDDEA30054912.
Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet. 2014;46:1327–32.
Wolking S, May P, Mei D, Møller RS, Balestrini S, Helbig KL, et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 2019;92:e1238–49.
Fujiwara T, Snada M, Kofuji T, Yoshikawa T, Akagawa K. HPC-1/syntaxin 1A gene knockout mice show abnormal behavior possibly related to a disruption in 5-HTergic systems. Eur J Neurosci. 2010;32:99–107.
Fujiwara T, Kofuji T, Akagawa K. Disturbance of the reciprocal-interaction between the OXTergic and DAergic systems in the CNS causes atypical social behavior in syntaxin 1A knockout mice. Behavioural Brain Res. 2021;413:113447.
Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C. MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum Mutat. 2019;40:1030–8.
Acknowledgements
We thank the families for their participation and support of this study.
Funding
FL and AG received funding from European Union and Région Normandie in the context of Recherche Innovation Normandie (RIN 2018). Europe gets involved in Normandie with the European Regional Development Fund (ERDF). Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: RAJ, KP. Investigation: JL, KP. Structural modeling: HS. Clinical data and genetic analysis: RAJ, FL, AG, KG, EA, SB, OK, CM, AV, A-SD-P, SJ. Visualization: HS, JL. Writing—original draft preparation: JL, KP, HS. Review and editing of manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study was approved by the ethics committee of the University of Leipzig, Germany (402/16-ek).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luppe, J., Sticht, H., Lecoquierre, F. et al. Heterozygous and homozygous variants in STX1A cause a neurodevelopmental disorder with or without epilepsy. Eur J Hum Genet 31, 345–352 (2023). https://doi.org/10.1038/s41431-022-01269-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01269-6
This article is cited by
-
2023 in the European Journal of Human Genetics
European Journal of Human Genetics (2024)
-
Studying ultra-rare variants in STX1A uncovers a novel neurodevelopmental disorder
European Journal of Human Genetics (2023)
-
Genes=disease (?)
European Journal of Human Genetics (2023)
