
Abstract
Pharmacoresistant epilepsy presenting during infancy poses both diagnostic and therapeutic challenges. We aim to identify diagnostic yield and treatment implications of exome sequencing (ES) as first-tier genetic testing for infantile-onset pharmacoresistant epilepsy. From June 2016 to December 2020, we enrolled patients with infantile-onset (age ≤ 12 months) pharmacoresistant epilepsy. 103 unrelated patients underwent ES. Clinical characteristics and changes in management due to the molecular diagnosis were studied. 42% (43/103) had epilepsy onset within the first month of life. After ES as first-tier genetic testing, 62% (64/103) of the cases were solved. Two partially solved cases (2%; 2/103) with heterozygous variants identified in ALDH7A1 known to cause autosomal recessive pyridoxine dependent epilepsy underwent genome sequencing (GS). Two novel large deletions in ALDH7A1 were detected in both cases. ES identified 66 pathogenic and likely pathogenic single nucleotide variants (SNVs) in 27 genes. 19 variants have not been previously reported. GS identified two additional copy number variations (CNVs). The most common disease-causing genes are SCN1A (13%; 13/103) and KCNQ2 (8%; 8/103). Eight percent (8/103) of the patients had treatable disorders and specific treatments were provided resulting in seizure freedom. Pyridoxine dependent epilepsy was the most common treatable epilepsy (6%; 6/103). Furthermore, 35% (36/103) had genetic defects which guided gene-specific treatments. Altogether, the diagnostic yield is 64%. Molecular diagnoses change management in 43% of the cases. This study substantiates the use of next generation sequencing (NGS) as the first-tier genetic investigation in infantile-onset pharmacoresistant epilepsy.
Similar content being viewed by others

Introduction
Infantile-onset pharmacoresistant epilepsy represents a significant subpopulation in pediatric epilepsy patients. The incidence of infantile-onset epilepsy is 0.8–1.2 per 1000 live births [1,2,3]. Refractory seizures presenting at this age usually result in developmental delay or arrest and various comorbidities. Patients with difficult-to-treat epileptic conditions have a poor quality of life and high mortality rate [4]. The clinical and genetic heterogeneity makes an accurate and specific diagnosis challenging. Acquired perinatal and postnatal brain injuries, and metabolic disorders are known etiologies in a proportion of infantile-onset epilepsy patients [5]. The remaining large proportion of the patients are known to have genetic defects. Copy number variations and chromosomal abnormalities account for 6-18% of the cases [6, 7]. It has been demonstrated that the most common genetic causes among infantile-onset epilepsy patients are monogenic disorders [8, 9].
Over the past decade, next generation sequencing (NGS) technology, including targeted NGS, exome sequencing (ES) and genome sequencing (GS) have led to an exponential increase in the diagnostic rate and the discovery of novel causative genes [9,10,11,12,13,14,15,16,17,18,19]. The diagnostic yield of NGS differs depending on the case inclusion and test methodology. Several studies employed multi-tiered tests, including biochemical testing, microarray and targeted NGS. Inclusion strategies regarding the perinatal history, developmental status, neuroimaging findings, previous biochemical and genetic testing vary in different studies [3, 12,13,14,15, 17, 20,21,22,23,24,25]. Although there have been several previous studies, a clear threshold to perform NGS on infants with epilepsy has rarely been addressed. Since the impact on treatments depends on the patients’ particular disorders, such as vitamin-responsive epilepsy, neurometabolic disorders and certain channelopathies [16, 26,27,28,29,30], identification of genes responsible for patients with infantile-onset pharmacoresistant epilepsy is crucial.
Here, we perform a study to unravel the underlying genetic causes in 103 unrelated patients with infantile-onset pharmacoresistant epilepsy using ES as the first-tier genetic testing.
Methods
Patient selection
From June 2016 to December 2020, patients with infantile-onset (seizure onset ≤12 months) pharmacoresistant epilepsy who were seen or referred for genetic testing at a tertiary care center (King Chulalongkorn Memorial Hospital) were recruited. Pharmacoresistant epilepsy is defined according to the International League Against Epilepsy (ILAE) as failure of adequate trials of two tolerated, appropriately chosen and used antiepileptic drugs (whether as monotherapies or combinations) to achieve sustained seizure freedom [31]. All patients were seen by pediatric neurologists and/or geneticists. The choice to add or discontinue the antiepileptic drugs in each patient was made by pediatric neurologists. Family history, physical examination, seizure semiologies, neuroimaging and electroencephalogram (EEG) and previous genetic testing data were collected. The neuroimaging results were reviewed by neuroradiologists. Patients with dysmorphic features consistent with known genetic syndromes and those with established genetic diagnosis were excluded.
This study was approved by the Institutional Review Board of Faculty of Medicine, Chulalongkorn University, Thailand (IRB No. 264/62). Written informed consent was obtained from parents or legal guardians of the participants.
Exome sequencing
After informed consent, three milliliters of peripheral blood were drawn from the patients and their available parents. Genomic DNA was extracted from peripheral blood leukocytes by using a Puregene blood kit (Qiagen, Hilden, Germany). The DNA samples were sent to Macrogen Inc., Seoul, Korea for exome sequencing. The libraries were enriched by SureSelect Human All Exon V5 kits and subsequently were sequenced using Illumina HiSeq 2000 Sequencer with a target output of 6 GB.
Short-read and long-read genome sequencing
The DNA samples were sent to Beijing Genomics Institute (BGI)., Beijing, China for short-read GS. For long-read Pacific Bioscience (PacBio) HiFi sequencing, single molecule, realtime (SMRT) bell libraries were prepared using the SMRTbell Express Template Prep Kit 2.0.
Variant identification
Sequence reads in FASTQ sequencing files were aligned to the Human Reference Genome hg19 from UCSC using Burrows-Wheeler Alignment (BWA) software (http://bio-bwa.sourceforge.net/). For any ES to be further analyzed, it was required to have 20-fold coverage in more than 95% of the targeted regions. After reads were aligned to the human reference genome (hg19) using BWA, the single nucleotide variants (SNVs) and small insertions/ deletions (indels) were detected by GATK Haplotypecaller and annotated by dbSNP&1000 G. We used GRIDSS, ERDS [32, 33] to detect structural variants (SVs) and copy number variants (CNVS) obtained from short-read GS data. For long-read PacBio HiFi GS data, read mapping, variant calling, and genome assembly were performed using a Snakemake workflow. HiFi reads were mapped using pbmm2 version 1.8.0 and SVs were called using pbsv 2.8.1. Single nucleotide variants (SNVs) were called using DeepVariant version 1.4.0 following DeepVariant best practices for PacBio reads.
Variant prioritization
A list of 728 genes associated with Genetic Epilepsy Syndrome according to Genomics England PanelApp (https://panelapp.genomicsengland.co.uk/panels/402/) was used for the first step of analysis. Both nuclear and mitochondrial genes known to be associated with epilepsy were included. The coding missense, nonsense, frameshift and splice site variants were first targeted. We also followed a variant prioritization using Exomiser (https://github.com/exomiser/Exomiser) to identify causative variants. HPO terms (HP:0001263; global developmental delay, HP:0200134; epileptic encephalopathy and HP:0001250; seizures) were used. If no variants within the gene list were identified, candidate pathogenic variants were then selected according to the following criteria (1) population frequency <1% in gnomAD (http://gnomad.broadinstitute.org/) and Thai reference exome database (T-REx) [34]. (2) variants predicted to have a functional impact on coding regions (predicted missense, nonsense, consensus donor/acceptor splice site mutations and insertions/deletions); (3) missense variants determined as damaging or disease-causing by at least 3 in silico predictive mutation impact software: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org/), M-CAP (http://bejerano.stanford.edu/mcap/), CADD (http://cadd.gs.washington.edu/). Variants were considered novel if they were not reported in the Genome Aggregation Database (gnomAD), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), PubMed literature, and not identified in our in-house Thai reference exome database (T-REx) [34]. Candidate variants were classified according to the recommendation of American College of Medical Genetics and Genomics (ACMG) [35, 36]. All candidate variants were also evaluated by clinical geneticists and neurologists.
Cases in which pathogenic or likely pathogenic variants in genes consistent with the phenotype were identified and zygosity matched the inheritance pattern were considered “solved”, while cases with only one heterozygous variant identified in a gene associated with recessive inheritance were considered “partially solved”. Partially solved cases underwent short-read genome sequencing to identify the second variant or other possible alterations. When negative, long-read Pacific Bioscience (PacBio) HiFi sequencing was subsequently performed. Unsolved cases were reanalyzed annually.
Statistical analysis
Direct associations between clinical characteristics (ie. family history, sex, epilepsy onset, seizure semiologies, EEG, neuroimaging findings, the number of antiepileptic drugs used, methods of ES analysis) and the presence of disease-causing variants were analyzed using the Pearson chi‐square test or Fisher’s exact test in case of small sample sizes in particular categories. The analyses were performed using SPSS Statistics version 26. A p-value of less than 0.05 is considered statistically significant.
Treatment implications
Genetic disorders with therapeutic implications are defined as conditions in which current literature supports a preferred medication or approach. These disorders include mutations in 16 epilepsy genes (ATP1A3, CDKL5, DEPDC5, GLUT1, GRIN2A, GRIN2B, KCNA2, KCNQ2, KCNT1, NPRL2, NPRL3, SCN1A, SCN2A, SCN8A, TSC1, TSC2) and all the treatable neurometabolic disorders [27,28,29,30].
Results
Clinical characteristics
One hundred and eight families with infantile-onset pharmacoresistant epilepsy from unrelated families were identified. Five patients with known dysmorphic syndromes or chromosomal disorders (ring chromosome 21, Miller-Dieker syndrome, 15q11q13 duplication syndrome, ring chromosome 14 mosaicism, Wolf-Hirschhorn syndrome) were excluded. The remaining 103 patients underwent ES.
Fifty-four percent (56/103) of the patients were male. 94% (97/103) of the patients were born at term. 93% (96/103) had no family history of epilepsy or developmental delay (Table 1). 42% (43/103) of the patients had seizures onset during the neonatal period. Of the 100 patients (97%; 100/103) with available neuroimaging data, 65 patients had unremarkable neuroimaging studies. The EEG data were available in 100 patients (97%). The common findings included multifocal epileptiform discharges (35%) and burst suppression pattern (25%). The number of antiepileptic drugs used before genetic testing ranged from 2 to 8. The clinical characteristics of the patients are summarized in Table 1. The clinical data of each patient are presented in Table S1.
Diagnostic yield and genetic findings
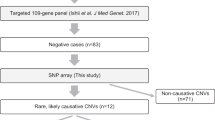
Of the 103 unrelated probands, 22, 79 and 2 families underwent ES using only one member (the proband; solo), three members (the proband and his/her parents; trio) and four members (the proband, his/her sibling and parents; Quadro), respectively. From the first ES analysis, 61 cases (59%) were solved and 2 cases (2%) with pyridoxine dependent epilepsy were partially solved. Both partially solved cases were further analyzed using short-read and long-read GS (Fig. 1). Reanalysis of ES of the unsolved cases led to molecular diagnoses in 3 more patients (7.5%; 3/40). ES revealed disease-causing variants in 64 patients (62%). 66 pathogenic and likely pathogenic SNVs were found in 27 genes known to be associated with epilepsy (Fig. 2A). 19 variants (29%) have never been previously reported. GS identified two novel pathogenic CNVs in partially solved cases. Three patients harbored variants of uncertain significance. Characteristics of all the variants are shown in Table S2. Confirmed and presumed (negative family history without parental sequencing) autosomal dominant de novo variants accounted for 71% (47/66) and confirmed X-linked de novo variants accounted for 8% (5/66) (Fig. 2C). The most common type was missense variants (Fig. 2D). The diagnostic rate was higher among patients with neonatal-onset (p-value = 0.048; Table S3).

Enrollment and allocation of the study population.

Categories of monogenic causes identified. The different shades of red pies represent cases with disease-causing variants identified. The blue portion represents unsolved cases. Of note, two patients (patients 1 and 2) with one likely pathogenic variant identified in the ALDH7A1 gene known to cause autosomal recessive pyridoxine dependent epilepsy are considered “partially solved” after ES (A). Percentage of patients with monogenic disorders and treatment implications (B). Specific treatments were administered and resulted in seizure freedom in patients with ALDH7A1 (n = 6), PNPO (n = 1) and BTD (n = 1) mutations. Molecular diagnosis that could guide treatment included variants identified in ATP1A3 (n = 3), PDHA1 (n = 1), KCNA2 (n = 1), KCNT1 (n = 3), KCNQ2 (n = 8), SCN1A (n = 13), SCN2A (n = 2), SCN8A (n = 4). Mode of inheritance (C). Types of disease-causing variants. The numbers following the semicolon (;) indicate the number of patients (D).
Treatment implications
Of all the patients enrolled, 8% (8/103) had treatable disorders and precision therapy resulted in seizure freedom. 35% (36/103) had genetic defects which guided gene-specific medications. Overall, 43% (44/103) of the patients harbored causative variants with treatment implications (Fig. 2B). The disease-causing variants identified in ALDH7A1 (n = 6), PNPO (n = 1), and BTD (n = 1), led to specific treatments and discontinuation of other antiepileptic drugs. All the patients were seizure-free after receiving precision therapy. The clinical data of the patients with identified variants in treatable disorders are summarized in Table 2. The disease-causing variants found in SCN1A (n = 13), SCN2A (n = 3), SCN8A (n = 4), ATP1A3 (n = 3), KCNA2, (n = 1), KCNT1 (n = 3), KCNQ2 (n = 8), and PDHA1 (n = 1) could guide treatment decisions [28].
The most common treatable disorder found in our cohort was the autosomal recessive pyridoxine-dependent epilepsy. Six patients harbored pathogenic and likely pathogenic variants in the ALDH7A1 gene (Table 2). Five of them had their first seizure during the neonatal period. Trial of pyridoxine (vitamin B6) and pyridoxal 5’-phosphate along with EEG monitoring was done in all six patients prior to genetic testing. Two out of six patients (patient 3 and 6) with ALDH7A1 variants did not show clear EEG responses. The biochemical testing including plasma and urinary alpha-aminoadipic semialdehyde (α-AASA), plasma and cerebral spinal fluid pipecolic acid and cerebrospinal fluid monoamine metabolites, were unavailable in our setting. ES successfully identified the disease-causing variants in four patients (Patients 3, 4, 5, 6). In patient 1, the previously reported c.1439 T > C (p.Ile480Thr) variant was found on the paternal allele. In patient 2, the previously reported c.1061 A > G (p.Tyr354Cys) variant was found on the maternal allele. Genome sequencing was performed in patients 1 and 2. In patient 1, short-read GS identified a novel 5,299-bp deletion (chr5:125914331-125919629del; GRCh37/hg19; NM_001182.5:c.388_518-1428del) encompassing the entire exon 5 of the ALDH7A1 gene in the maternal allele. In patient 2, long-read GS identified a novel 53,417-bp deletion (chr5:125903412-125956829del; NM_001182.4:c.−25939_871 + 539del) encompassing exons 1-9 of the ALDH7A1 gene and exons 1-4 of the PHAX gene in the paternal allele (Fig. 3).

A Intragenic deletions in the ALDH7A1 gene found in patients with pyridoxine dependent epilepsy. The black bars depict all previously reported deletions (Kanno et al. 2007; exon 17, Plecko et al. 2007; exon 7, Perez et al. 2003; exons 12-18, Mefford et al. 2015; exon 7, exons 8-9, exons 14-17, exon 8 and flanking exons 1-9 of the GRAMD3 gene). The red bars depict the deletions found in patients 1 and 2 in this study. The yellow and green bars represent the flanking sequence in the 5’ and 3’ regions, respectively. B Binary alignment map (BAM) and FASQ capture from whole genome sequencing data. The horizontal red bars represent the 5299-bp deletion found in patient 1 and his mother and the 53,417-bp deletion found in patient 2 and his father. The DNA sequences flanking the deletions are highlighted in green for the 5’ region and yellow for the 3’ region.
Interestingly, four unrelated patients with pyridoxine dependent epilepsy harbored the c.1061 A > G (p. Tyr354Cys) variant and two unrelated patients harbored the c.1547 A > G (p.Tyr516Cys) variant. Further studies could help clarify whether these recurrent variants are due to a founder effect.
Discussion
In this study, ES was performed in 103 patients with infantile-onset pharmacoresistant epilepsy. As a first-tier diagnostic tool, ES solved 62% (64/103) of the cases. Pathogenic and likely pathogenic SNVs were identified in 66 patients. 29% (19/66) of the identified variants have not been previously described. GS identified two CNVs and solved additional 2% (2/103) of the cases. In total, 64% of the cases were solved. The SCN1A gene was found to be responsible for the majority of cases (13%; 13/103). This was similar to previous large cohort studies of pediatric epilepsy [14, 22, 37]. Mutations in KCNQ2 were the most common among neonatal-onset epilepsy and patients with burst suppression patterns on EEG (Table S3). Eight percent (8/103) of all patients had neurometabolic disorders with specific treatments that resulted in seizure freedom. In addition, 35% (36/103) had molecular diagnoses that could guide treatment decisions. Overall, 43% of the patients harbored variants with treatment implications. Our study is the first and largest to investigate the genetic defects underlying infantile-onset pharmacoresistant epilepsy in the Thai population.
Several factors contribute to the relatively high diagnostic yield in this study. The inclusion of patients with early age of onset poses a strong genetic potential. This study was performed at a tertiary care center. Exclusion of all possible acquired causes of seizures was usually done in most patients prior to referral. Furthermore, the majority (77%; 79/103) of the patients underwent trio-NGS analysis. Parents-proband trio analysis facilitates the identification of de novo variants which account for most of the disease-causing variants in severe genetic epilepsy [38]. Although there is no statistical significance between the diagnostic yield of solo, trio and quadro analysis in this study, there is a trend of higher yield for trio compared to solo (Table 1). The relatively small sample size might contribute to the non-statistical significance. Previous studies have shown the relatively high diagnostic efficacy of trio-analysis, particularly in neurodevelopmental disorders [39, 40].
The diagnostic rate of patients with neonatal-onset epilepsy was higher than those with later onset (74% VS 57%), but the difference did not reach statistical significance (p-value=0.54). When diagnostic rates were compared between patients with epilepsy onset at different months, those with neonatal-onset epilepsy had a significantly higher diagnostic rate (p-value = 0.048; Table S3). The other clinical characteristics of patients, including seizure semiologies, EEG findings, neuroimaging findings and the total number of antiepileptic drugs used before testing, were unrelated to the variants identified. These findings suggested that patient characteristics in infantile-onset pharmacoresistant epilepsy could not be used to predict the likelihood of identifying disease-associated variants. Therefore, ES should be considered in all patients with infantile-onset pharmacoresistant epilepsy, especially in neonates.
Although data have shown a comparable utility and cost-effectiveness between targeted gene panel and ES, this study employed the use of ES. The purpose of this study was also to map out the genetic landscape of infantile-onset epilepsy in the Thai population. Since there have been no previous studies, choosing the most rational gene panel for the Thai population could be challenging and may confine our knowledge to a certain gene list. However, the result of this study revealed that the genetic landscape of infant epilepsy in the Thai population was similar to other populations. Our findings suggested a targeted gene panel could also be employed in the Thai population with a potentially high diagnostic yield for clinical settings.
Another established benefit of ES is the feasibility of reanalysis. Although, most of the cases (59%; 61/103) in this cohort were diagnosed at the initial ES analysis. Reanalysis of the initially negative trio-ES revealed the diagnosis in three more patients with de novo pathogenic variants in newly identified epilepsy genes (PHACTR1, GABRA5 and PACS2). The value of periodic reanalysis of ES has also been established in previous studies [41, 42].
Previous studies showed varying yields of treatable disorders ranging from 4.5 to 6%, depending on case inclusion, previous diagnostic studies and the number of genes tested [13, 22]. This study is the first using ES as a first-tier genetic test in patients who need two or more antiepileptic drugs. In this cohort, 8% of the patients had treatable disorders which include pyridoxine (B6) dependent epilepsy, pyridoxamine 5’-phosphate (P5’P) oxidase deficiency and biotinidase deficiency. The most common treatable disorder in this cohort is pyridoxine dependent epilepsy. Four patients in this cohort showed EEG responses after IV-B6 and two patients did not (Table 2). Previous studies have shown that the EEG responses to IV-B6 neither identify nor exclude pyridoxine-dependent epilepsy. In order to safely wean off other antiepileptic drugs, diagnosis should be established using biochemical and/or DNA analysis [43]. Biochemical testing for various neurological disorders with epilepsy was not available in our setting as well as in many other hospitals. Given the wide range of neurometabolic conditions that can cause epilepsy, selecting and interpreting biochemical tests can be challenging and time-consuming [29, 30]. This study provided evidence suggesting that the molecular approach could also lead to the diagnosis.
In this study, short-read and long-read GS were done in two partially solved cases with autosomal recessive pyridoxine dependent epilepsy. Short-read GS identified a novel 5,299-bp deletion encompassing the entire exon 5 of the ALDH7A1 gene in patient 1 and his mother. Long-read GS identified a novel 53,417-bp deletion encompassing exons 1-9 of the ALDH7A1 gene and exons 1-4 of the PHAX gene in patient 2 and his father (Fig. 3). The predicted effect of this partial PHAX gene deletion is unclear. A monoallelic deletion of the PHAX gene is less likely to be sufficient to cause an epileptic or neurodevelopmental phenotype since patient 2 inherited the deletion from an asymptomatic father. Mefford et al. revealed that deletions involving exons within the ALDH7A1 gene accounted for the majority of mutations in patients with pyridoxine dependent epilepsy who harbored only one documented SNV. To our knowledge, there have been 8 previously reported intragenic deletions in the ALDH7A1 gene (Fig. 3) [44,45,46,47]. The deletion involving the flanking PHAX gene is first described here. This study emphasizes that epilepsy patients with one identifiable pathogenic variant in the ALDH7A1 gene warrant further testing for copy number changes involving the ALHD7A1 and nearby genomic regions. We also propose the use of GS in partially solved cases. Further studies are required to address the diagnostic yield and the cost-effectiveness of the strategy.
Four of the eight patients with treatable disorders who received the diagnosis later in the clinical course had a dismal developmental outcome. Patient 7, who had pyridoxamine 5’-phosphate oxidase deficiency, experienced super-refractory status epilepticus after the conventional dose of pyridoxamine 5’ phosphate (P5’P). Only when the genetic result was available that the P5’P dosage was increased to achieve seizure control. This emphasizes the need for a rapid diagnosis. Accurate and timely administration of medications can potentially improve long-term outcomes. With declining costs, ES is an effective diagnostic tool for clinical diagnosis. Growing evidence supports the clinical utility and cost-effectiveness of rapid ES and WGS in critically-ill pediatric patients including patients with seizures [48,49,50]. Our study provided evidence strongly supporting early use of ES in patients with infantile-onset pharmacoresistant epilepsy.
In summary, ES led to molecular diagnosis in 62% (64/103) of cases. Of all the 66 variants found to be responsible for the diseases, 19 (29%) were novel. Short-read and long-read GS identified two large deletions and solved additional 2% (2/103) of the cases. In total, 64% of the cases were solved. This study is the first and largest to investigate genetic causes in infantile-onset pharmacoresistant epilepsy in the Thai population and suggests the use of ES as the first-tier genetic testing to increase diagnostic yield and improve treatment outcomes. Overall, 43% (44/103) of the patients in this study have monogenic disorders with treatment implications.
Data availability
The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Symonds JD, Zuberi SM, Stewart K, McLellan A, O’Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142:2303–18.
Gaily E, Lommi M, Lapatto R, Lehesjoki AE. Incidence and outcome of epilepsy syndromes with onset in the first year of life: A retrospective population-based study. Epilepsia. 2016;57:1594–601.
Stödberg T, Tomson T, Barbaro M, Stranneheim H, Anderlid BM, Carlsson S, et al. Epilepsy syndromes, etiologies, and the use of next-generation sequencing in epilepsy presenting in the first 2 years of life: A population-based study. Epilepsia. 2020;61:2486–99.
Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014;37:59–70.
Eltze CM, Chong WK, Cox T, Whitney A, Cortina-Borja M, Chin RF, et al. A population-based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54:437–45.
Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010;51:676–85.
Allen NM, Conroy J, Shahwan A, Ennis S, Lynch B, Lynch SA, et al. Chromosomal microarray in unexplained severe early onset epilepsy - A single centre cohort. Eur J Paediatr Neurol. 2015;19:390–4.
Butler KM, da Silva C, Alexander JJ, Hegde M, Escayg A. Diagnostic Yield From 339 Epilepsy Patients Screened on a Clinical Gene Panel. Pediatr Neurol. 2017;77:61–66.
Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia. 2016;57:e12–17.
Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018;17:e12456.
Olson HE, Kelly M, LaCoursiere CM, Pinsky R, Tambunan D, Shain C, et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol. 2017;81:419–29.
Segal E, Pedro H, Valdez-Gonzalez K, Parisotto S, Gliksman F, Thompson S, et al. Diagnostic Yield of Epilepsy Panels in Children With Medication-Refractory Epilepsy. Pediatr Neurol. 2016;64:66–71.
Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–16.
Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie H, et al. Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/Developmental Disabilities. PLoS One. 2015;10:e0141782.
Ostrander BEP, Butterfield RJ, Pedersen BS, Farrell AJ, Layer RM, Ward A, et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom Med. 2018;3:22.
Demos M, Guella I, DeGuzman C, McKenzie MB, Buerki SE, Evans DM, et al. Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front Neurol. 2019;10:434.
Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53:1387–98.
Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53:310–7.
Ortega-Moreno L, Giraldez BG, Soto-Insuga V, Losada-Del Pozo R, Rodrigo-Moreno M, Alarcon-Morcillo C, et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS One. 2017;12:e0188978.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–30.
Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, et al. The genetic landscape of infantile spasms. Hum Mol Genet. 2014;23:4846–58.
Costain G, Cordeiro D, Matviychuk D, Mercimek-Andrews S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience. 2019;418:291–310.
Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, et al. Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Res. 2018;141:48–55.
Kothur K, Holman K, Farnsworth E, Ho G, Lorentzos M, Troedson C, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure. 2018;59:132–40.
Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. 2019;60:155–64.
Hoelz H, Herdl C, Gerstl L, Tacke M, Vill K, von Stuelpnagel C, et al. Impact on Clinical Decision Making of Next-Generation Sequencing in Pediatric Epilepsy in a Tertiary Epilepsy Referral Center. Clin EEG Neurosci. 2020;51:61–69.
Johannessen Landmark C, Potschka H, Auvin S, Wilmshurst JM, Johannessen SI, Kasteleijn-Nolst Trenite D, et al. The role of new medical treatments for the management of developmental and epileptic encephalopathies: Novel concepts and results. Epilepsia. 2021.
Devinsky O, Vezzani A, O’Brien TJ, Jette N, Scheffer IE, de Curtis M, et al. Epilepsy. Nat Rev Dis Prim. 2018;4:18024.
Almannai M, El-Hattab AW. Inborn Errors of Metabolism with Seizures: Defects of Glycine and Serine Metabolism and Cofactor-Related Disorders. Pediatr Clin North Am. 2018;65:279–99.
Sharma S, Prasad AN. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int J Mol Sci. 2017;18:1384.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–77.
Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20:117.
Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91:408–21.
Shotelersuk V, Wichadakul D, Ngamphiw C, Srichomthong C, Phokaew C, Wilantho A, et al. The Thai reference exome (T-REx) variant database. Clin Genet. 2021;100:703–12.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245–57.
Arafat A, Jing P, Ma Y, Pu M, Nan G, Fang H, et al. Unexplained Early Infantile Epileptic Encephalopathy in Han Chinese Children: Next-Generation Sequencing and Phenotype Enriching. Sci Rep. 2017;7:46227.
Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet. 2017;101:664–85.
Tan TY, Lunke S, Chong B, Phelan D, Fanjul-Fernandez M, Marum JE, et al. A head-to-head evaluation of the diagnostic efficacy and costs of trio versus singleton exome sequencing analysis. Eur J Hum Genet. 2019;27:1791–9.
Gao C, Wang X, Mei S, Li D, Duan J, Zhang P, et al. Diagnostic Yields of Trio-WES Accompanied by CNVseq for Rare Neurodevelopmental Disorders. Front Genet. 2019;10:485.
Li J, Gao K, Yan H, Xiangwei W, Liu N, Wang T, et al. Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene. 2019;700:168–75.
Rochtus A, Olson HE, Smith L, Keith LG, El Achkar C, Taylor A, et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia. 2020;61:249–58.
Bok LA, Maurits NM, Willemsen MA, Jakobs C, Teune LK, Poll-The BT, et al. The EEG response to pyridoxine-IV neither identifies nor excludes pyridoxine-dependent epilepsy. Epilepsia. 2010;51:2406–11.
Kanno J, Kure S, Narisawa A, Kamada F, Takayanagi M, Yamamoto K, et al. Allelic and non-allelic heterogeneities in pyridoxine dependent seizures revealed by ALDH7A1 mutational analysis. Mol Genet Metab. 2007;91:384–9.
Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E, Jakobs C, et al. Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat. 2007;28:19–26.
Mefford HC, Zemel M, Geraghty E, Cook J, Clayton PT, Paul K, et al. Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination. Neurology. 2015;85:756–62.
Perez B, Gutierrez-Solana LG, Verdu A, Merinero B, Yuste-Checa P, Ruiz-Sala P, et al. Clinical, biochemical, and molecular studies in pyridoxine-dependent epilepsy. Antisense therapy as possible new therapeutic option. Epilepsia. 2013;54:239–48.
Clark MM, Hildreth A, Batalov S, Ding Y, Chowdhury S, Watkins K, et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med 2019;11:eaat6177.
Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10.
Kamolvisit W, Phowthongkum P, Boonsimma P, Kuptanon C, Rojnueangnit K, Wattanasirichaigoon D, et al. Rapid exome sequencing as the first-tier investigation for diagnosis of acutely and severely ill children and adults in Thailand. Clin Genet. 2021;100:100–5.
Acknowledgements
We would like to thank the patients and their families for participating in this study. We also thank Natrujee Wiwattanadittakul M.D., Chinnuwat Sanguansermsri M.D., Kamornwan Katanyuwong M.D., Division of Neurology, Department of Pediatrics, Maharaj Nakorn Chiang Mai Hospital; Somjit Sri-Udomkajorn M.D., Thanin Wechapinan M.D., Sirorat Suwannachote M.D., Chulaluck Kuptanon M.D., Department of Pediatrics, Queen Sirikit National Institute of Child Health, Surachai Likasitwattankul, M.D., Sorawit Viravan, M.D., Mongkon Chanvanichtrakool, M.D. Department of Pediatrics, Siriraj Hospital; Thitiporn Fangsaad, M.D. Department of Pediatrics, Bhumibol Adulyadej Hospital; Vitchayaporn Emarach Saengow, M.D. Department of Pediatrics, Maharat Nakhon Ratchasima Hospital, for their excellent care of the patients.
Funding
The study was supported by Health Systems Research Institute (65-040, 64-125).
Author information
Authors and Affiliations
Contributions
PB, WK, SP and TD collected data. WC, CP, CI, and RI performed DNA sequencing and analysis. PB and CI analyzed the data, drafted and revised the paper. KS and VS revised the draft. KS and VS conceived the study and obtained funding. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Institutional Review Board of Faculty of Medicine, Chulalongkorn University, Thailand (IRB No. 264/62). Written informed consent was obtained from parents or legal guardians of the participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Boonsimma, P., Ittiwut, C., Kamolvisit, W. et al. Exome sequencing as first-tier genetic testing in infantile-onset pharmacoresistant epilepsy: diagnostic yield and treatment impact. Eur J Hum Genet 31, 179–187 (2023). https://doi.org/10.1038/s41431-022-01202-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01202-x
This article is cited by
-
The value of exomes across the ages
European Journal of Human Genetics (2023)
-
Nine patients with KCNQ2-related neonatal seizures and functional studies of two missense variants
Scientific Reports (2023)
