
Abstract
In genetic studies of psychiatric disorders in the pre-genome-wide association study (GWAS) era, one of the most commonly studied loci is the serotonin transporter (SLC6A4) promoter polymorphism, a 43-base-pair insertion/deletion polymorphism in the promoter region (5-HTTLPR). The genetic association signals between 5-HTTLPR and psychiatric phenotypes, however, have been inconsistent across many studies. Since the polymorphism cannot be tested via available SNP arrays, we had previously proposed an efficient machine learning algorithm to predict the genotypes of 5-HTTLPR based on the genotypes of eight nearby SNPs, which requires access to individual-level genotype and phenotype data. To utilize the advantage of publicly available GWAS summary statistics obtained from studies with very large sample sizes, we develop a GWAS summary-statistics-based approach for testing the variable number of tandem repeat (VNTR) associations with various phenotypes. We first cross-verify the accuracy of the summary-statistics-based approach for 61 phenotypes in the UK Biobank. Since we observed a strong similarity between the predicted individual-level 5-HTTLPR genotype-based approach and the summary-statistics-based approach, we applied our method to the available neurobehavioral GWAS summary statistics data obtained from large-scale GWAS. We found no genome-wide significant evidence for association between 5-HTTLPR and any of the neurobehavioral traits. We did observe, however, genome-wide significant evidence for association between this locus and human adult height, BMI, and total cholesterol. Our summary-statistics-based approach provides a systematic way to examine the role of VNTRs and related types of genetic polymorphisms in disease risk and trait susceptibility of phenotypes for which large-scale GWAS summary statistics data are available.
Similar content being viewed by others


Introduction
Large-scale human genetic studies have resulted in major breakthroughs for our understanding of the genetic architecture of complex human traits. SNP arrays for genome-wide association studies (GWAS) in up to millions of subjects are extensively used for the detection of common risk alleles for polygenic disorders such as schizophrenia [1] and bipolar disorder [2]. On the other hand, the approach of high-throughput sequencing in tens of thousands of individuals has identified rare and deleterious sequence variants contributing to neurodevelopmental disorders such as autism spectrum disorder [3] and schizophrenia [4]. One type of genetic variation that was extensively studied before the advances in genomic technology, the variable number of tandem repeats (VNTRs) class of variants [5], has eluded further large-scale analysis in the genomic era. SNP arrays allow for the high-throughput collection of single nucleotide variants but not for VNTRs, microsatellite polymorphisms, and related genetic variants that are also abundant in the human genome.
In genetic studies of psychiatric disorders, one of the most commonly studied locus in the pre-GWAS era [6], is the serotonin transporter (5-HTT or SLC6A4) promoter polymorphism, a 43-base-pair insertion/deletion polymorphism in the promoter region (5-HTTLPR) with a long (L) and a short (S) allele. The SLC6A4 gene was long considered a strong candidate gene for neurobehavioral traits and disorders. The reason is that many antidepressant drugs were reported to selectively inhibit the function of the serotonin transporter (5-HTT or SLC6A4), the gene product of which mediates the reuptake of monoamine serotonin (5-HT), a key neurotransmitter in the brain. Consequently, genetic variants such as the 5-HTTLPR were considered important factors to modulate the expression of the serotonin transporter protein under certain physiologic conditions. However, the genetic association signal findings between 5-HTTLPR and psychiatric and neurobehavioral phenotypes have been found to be inconsistent across studies, few indicating an association, for example, when interacting with stressful life events [7], but a meta-analysis with a total sample size of more than 14,000 participants failed to provide any evidence for association with depression with or without stressful life events [8]. Another systematic gene x environment interaction (GxE) meta-analysis of this locus showed that the published studies are underpowered and that the main effects of 5-HTTLPR genotype and the interaction effect of stressful life events on risk of depression are negligible [9]. Another meta-analysis of 54 studies that included 40,749 subjects, in which environmental effects as well as phenotype outcomes were taken very broadly, reported a positive GxE effect with the S allele associated with increased stress sensitivity [10].
Since the 5-HTTLPR VNTR cannot be tested via available SNP arrays, we previously proposed an efficient machine learning algorithm to predict the genotypes of 5-HTTLPR based on genotypes of eight nearby SNPs [11]. The predicted VNTR genotypes can then be used for association testing with a phenotype of interest. However, this approach requires access to individual-level genotype data for the eight SNPs and phenotype data; hence, it cannot utilize the advantage of publicly available GWAS summary statistics obtained from studies with very large sample sizes across various neurobehavioral phenotypes. In order to revisit the 5-HTTLPR for genetic analysis in the largest study samples available, we developed and applied a GWAS summary-statistics-based approach for association testing of this locus.
In short, in a training dataset that has PCR-based genotypes for 5-HTTLPR and the eight SNPs considered for our machine learning algorithm [11], we fit a linear model to estimate the allele dosage of 5-HTTLPR based on the eight SNPs. We use this prediction model to impute the allele dosage of 5-HTTLPR in a GWAS dataset (where 5-HTTLPR genotypes are not available) and perform association testing with the phenotype. We devise a summary-statistics-based version of this two-step procedure in which we only need the GWAS summary statistics at the eight SNPs for the phenotype. We first cross-verify the accuracy of the summary-statistics-based approach for 61 phenotypes in UK Biobank (UKBB). We contrast the association statistics computed by the summary-statistics-based approach and our previous genotype-based method. Since we observed a strong similarity between the two approaches, we applied our method to the available neurobehavioral GWAS summary statistics data with large sample size as well as of other disorders and traits. We found no genome-wide significant evidence for association between the 5-HTTLPR and any of the neurobehavioral traits. We did observe, however, genome-wide significant evidence for association between this locus and human adult height, body mass index (BMI), and total cholesterol. Our approach provides a systematic way to re-assess and examine the role of specific VNTRs and related types of genetic polymorphisms in disease risk and trait susceptibility of phenotypes for which summary-level data from large-scale GWAS are available.
Methods
Prediction of 5-HTTLPR genotypes based on tag SNPs
In an ideal scenario, if the genotype data of 5-HTTLPR is available along with the phenotype of interest in a sufficiently large study sample, we can directly assess the association between 5-HTTLPR and the phenotype. The PCR-based assay to genotype the serotonin transporter promoter polymorphism has suffered from technical difficulties as demonstrated by the lack of Hardy–Weinberg equilibrium in genotype data for this locus [9]. With the rise of use of SNP genotyping arrays, we previously proposed a method to predict the 5-HTTLPR VNTR genotypes based on nearby tag SNPs, which also requires the individual-level genotype data of the tag SNPs. We now move one step further, beyond the need to access individual-level genotype data but to leverage the largest available sample sizes for genetic studies used for GWAS. Our approach to examine the 5-HTTLPR VNTR is similar to the summary-statistics-based method proposed by Gusev et al. [12] to evaluate the association between the genetic component of a gene’s expression and a phenotype.
The serotonin transporter (SLC6A4) promoter polymorphism is a tandem repeat polymorphism, also called the 5-HTTLPR, with two alleles: one long allele (L) and one short allele (S), hence the three possible genotypes SS, SL, and LL. Using additive coding, i.e., counting the number of L alleles in a genotype, the coded genotype can take three possible values: 0, 1, 2. Suppose, we have a reference dataset containing the genotype data of 5-HTTLPR and the surrounding eight SNPs tagging 5-HTTLPR (Table 1), which we considered in our previous work [11]. We use an individual-level genotype dataset for these nine variants (5-HTTLPR and eight tag SNPs) that consists of 276 Dutch individuals comprising 126 normal individuals and 150 individuals diagnosed with schizophrenia [11]. We consider a linear regression model to estimate the component of the 5-HTTLPR genotypes that can be predicted by the eight tag SNPs.
Here g denotes the genotype of 5-HTTLPR, and xj denotes the genotype of jth tag SNP, j = 1,…,8. Let γj denote the regression coefficient corresponding to jth tag SNP’s genotype. We consider the genotype vector of each tag SNP to be normalized (zero mean and unity variance). We note that the predicted component of 5-HTTLPR genotypes, \(\hat g\), obtained from the linear regression can be continuous. The least square estimation of γ1,…,γ8 does not depend on the distribution of the noise ϵ. Since the sample size of the Dutch dataset (n = 276) is much larger than the number of tag SNPs, a linear regression model is adequate for model fitting instead of a penalized regression.
Let X denote the genotype data matrix for the eight tag SNPs (Table 1). Using the multiple linear regression model stated above, we obtain the least square estimate (LSE) of γ = (γ1,…,γ8)’ as: \(\hat \gamma = \left( {X^{\prime}X} \right)^{ - 1}X^{\prime}G\), where G is the genotype vector for 5-HTTLPR. Once we estimate γ, it is considered fixed in the down-stream analysis. If the 5-HTTLPR genotypes are missing in a different dataset, but the genotypes of the eight tag SNPs are available, we can use \(\widehat G = \widehat \gamma x\) to predict the 5-HTTLPR genotypes, where x is the genotype vector for the tag SNPs.
Evaluating association between 5-HTTLPR and a phenotype
Suppose, for n unrelated individuals in a new dataset, Y denotes the phenotype of interest, G denotes the genotype of 5-HTTLPR that is unobserved. We assume that the individual-level data for Y and genotypes of the tag SNPs are available, but the 5-HTTLPR genotypes are missing. Consider a linear regression of Y on unobserved G as follows:
Let X denote the genotype data matrix for the eight tag SNPs. Since G is unobserved, we can use our previously proposed machine learning method [11] to predict G based on X, and perform a test for association between predicted G and Y.
When individual-level data of Y and genotype data of the tag SNPs are also not available, we propose the following summary-statistics-based approach. We use the prediction model that was obtained from linear regression fitted in the Dutch reference dataset (Eq. 1) to predict the genotypes of 5-HTTLPR as: \(\hat G = X\hat \gamma\). Thus, we consider \(Y = \hat G\alpha + e\), where e is the random error. In this paper, we assume that \(\widehat \gamma\) is the same between the Dutch and other European populations. Next, we obtain the LSE of α in the linear regression \(Y = \hat G\alpha + e\).
Here, V is the LD matrix for the eight tag SNPs. We derive the above formula using the facts that, \(\hat G = X\hat \gamma\) and X’X = nV, since the genotype vector for each tag SNP is normalized. Suppose, Y is also separately regressed on the genotype of jth tag SNP as follows:
where Y and Xj are normalized. Let βj denote the marginal GWAS effect size of jth tag SNP on Y. LSE of βj is given by: \(\hat \beta _j = ( {X^{\prime}_jX_j} )^{ - 1}X^{\prime}Y = \frac{1}{n}X^{\prime}Y \Rightarrow X^{\prime}Y = n\hat \beta _j\). Since Xj is normalized, \(X^{\prime}_jX_j = n\). Denote the vector of estimated marginal GWAS effect size across the eight tag SNPs as: \(\hat \beta = ( {\hat \beta _1, \ldots ,\hat \beta _8} )^\prime\). Hence, for the tag SNPs considered together: \(X^{\prime}Y = n\hat \beta\). Thus, we can rewrite the expression of LSE of the effect size of 5-HTTLPR on Y, \(\hat \alpha\) (Eq. 3), as follows:
Next, we derive the expression of standard error of \(\hat \alpha\). Let us define the constant \(K_{wv} = \hat \gamma^{\prime}V\hat \gamma\). Hence,
Here, \({\mathop{{{{{{{\rm{cov}}}}}}}}} ( {\hat \beta } ) = \frac{{\sigma _y^2}}{n}V\) is the variance-covariance matrix of \(\hat \beta\), where \(\sigma _y^2 = {\mathop{{{{{{{\rm{var}}}}}}}}} \left( Y \right)\) and V is the LD matrix of the tag SNPs. So,
In practice, Y is normalized before running a GWAS. Hence, we assume that \(\sigma _y^2 = 1\). Suppose Zβ be the vector of marginal z-scores of associations between Y and the tag SNPs. We can rewrite the expression of Z-statistics for testing α = 0 as follows:
For a sufficiently large sample size, each element of Zβ follows a normal distribution. Hence, Zα also follows a normal distribution asymptotically. If Zα = z, a p value of association between 5-HTTLPR and Y can be obtained as P(U > z2), where U follows a χ2 distribution with one degree of freedom.
Thus, given \(Z_\beta ,\gamma ,V\), we can test α = 0. Therefore, using summary-level data on genetic association between a phenotype Y and the tag SNPs of 5-HTTLPR, the estimated prediction model for 5-HTTLPR genotypes (based on the Dutch reference dataset), and an estimate of the LD matrix of the tag SNPs, we can test for association between 5-HTTLPR and Y. We do not require any individual-level phenotype data and genotype data of 5-HTTLPR and related tag SNPs. We can use an estimate of V, the LD matrix of the tag SNPs, obtained based on the European individuals in 1000 genome data. To further increase the number of individuals in the LD reference panel, we combined the 276 Dutch individuals in our reference dataset with the European individuals in 1000 genome. We also provide the LD between 5-HTTLPR and its tag SNPs in Table 1. In the rest of this paper, we refer to our summary-statistics-based approach as VNTR.s.
Validation of summary-statistics-based approach in UK Biobank
In order to cross-verify the accuracy of the summary-statistics-based approach (VNTR.s), we conducted a phenome-wide association study for 5-HTTLPR and 61 phenotypes in the UKBB based on both of the individual-level prediction of 5-HTTLPR genotypes [11] and the summary-statistics-based approach VNTR.s. We contrast the results of association analyses obtained by these two methods to assess the accuracy of VNTR.s. We discuss the procedure of analyzing a single phenotype in UKBB (e.g., height).
First, we implement our previous machine learning approach (vertex discriminant analysis [11]) for 287,969 white British individuals in UKBB. We used the individual-level genotype data of the tag SNPs (Table 1) to predict 5-HTTLPR genotypes for the UKBB individuals. In our previous work [11], we estimated the coefficients of the 5-HTTLPR genotype prediction model based on the Finn dataset in which individual-level genotype data for both 5-HTTLPR and the tag SNPs were available for 2147 normal participants [11]. We used the same coefficients (Supplementary Table S3) to predict the 5-HTTLPR genotypes in UKBB. Next, we perform a linear regression of the phenotype on the predicted 5-HTTLPR genotypes to compute a p value of association while adjusting for relevant covariates, such as, age, sex, and top 20 principal components (PCs) of genetic ancestry. We refer to this approach as VNTR.g.
Next, we apply VNTR.s for the same phenotype in UKBB. We perform a linear regression of the phenotype on the genotype of each tag SNP of 5-HTTLPR, while adjusting for the same set of covariates, age, sex, and top 20 genetic ancestry of PCs. We obtain the summary statistics of association between the phenotype and each tag SNP from these regressions. We now assume that only the summary statistics for each tag SNP are available. Using the summary statistics and estimate of the LD matrix for the tag SNPs (as described above), we apply VNTR.s to compute the p value of association between 5-HTTLPR and the phenotype.
We applied VNTR.g and VNTR.s for 61 phenotypes in UKBB to compute the p values of association along with z-scores. We note that the genotype data for one of the tag SNPs, rs1061342 on chromosome 17 (Table 1), was missing from the UKBB individual-level genotype dataset available to us. Albeit under this limitation, we implemented VNTR.g and VNTR.s based on the remaining seven tag SNPs (Table 1). In our previous work [11], we evaluated the relative contribution of the tag SNPs to the prediction accuracy of the 5-HTTLPR genotypes with respect to partial and cumulative R2. The partial R2 for rs1061342, the missing SNP in UKBB, was very modest (Figure 1 in Lu et al. [11]) and the cumulative R2 increased from 0.83 to 0.84 after adding rs1061342 to the prediction model (sixth row of Table 2 in Lu et al. [11]). The total cumulative R2 of the full prediction model was 0.85. Thus, from the viewpoint of a stepwise regression, the prediction accuracy gets saturated before rs1061342 is added to the prediction model. Therefore, we emphasize that the comparison between VNTR.g and VNTR.s remains meaningful, albeit under the limitation of our UKBB individual-level genotype dataset.
Next, we contrast the results obtained by VNTR.g and VNTR.s for 61 UKBB phenotypes. Encouragingly, the p values obtained by the two approaches are very similar (Table 2). For example, for FEV1–FVC ratio, VNTR.g produced a p value of 1.6 × 10–8 and VNTR.s produced a p value of 3.2 × 10–8 (Table 2). For Albumin, VNTR.g produced a p value of 0.95 and VNTR.s produced a p value of 0.93 (Table 2). In the following, we provide an overall summary of the similarity.
For a UKBB phenotype, let P.g and P.s denote the p values obtained by VNTR.g and VNTR.s, respectively. Let y and x denote –log10(P.g) and –log10(P.s), respectively. Pearson’s correlation coefficient between y and x based on 61 UKBB phenotypes was estimated as 0.98. Spearman’s rank correlation coefficient was estimated to be 0.9. If we fit a linear regression of y on x, we obtain the following regression equation: y = –0.16 + 1.15x. Here, the slope regression coefficient was estimated as 1.15 with a standard error of 0.03, and the intercept coefficient was estimated to be –0.16 with a standard error of 0.07. Similarly, we regressed x on y and obtained the following regression equation: x = 0.19 + 0.83y. We also performed the non-parametric Kolmogorov–Smirnov (KS) test to evaluate if x and y have the same distribution. The KS test p value was 0.99 that strongly implies that x and y have the same underlying distribution. A plot of the observed values of x and y (Fig. 1) displays good concordance between the results obtained by VNTR.g and VNTR.s.

If y denotes –log10(p value) for VNTR.g and x denotes –log10(p value) for VNTR.s, the estimated linear regression equations were obtained as: y = –0.16 + 1.15x and x = 0.19 + 0.83y.
Thus, the overall analysis of 61 UKBB phenotypes demonstrates an excellent agreement between the outputs of VNTR.g and VNTR.s. This validates the use of VNTR.s for testing the association between 5-HTTLPR and a phenotype of interest based on publicly available summary-level data for association between the tag SNPs and the phenotype.
Application of the summary-statistics-based approach to various phenotypes
Using publicly available summary statistics data from GWAS with large sample sizes, we implemented VNTR.s to study association of 5-HTTLPR with a range of phenotypes mainly focusing on psychiatric and neurobehavioral phenotypes. We applied VNTR.s to nine psychiatric phenotypes in the psychiatric genetics consortium (PGC). We then examined 131 phenotypes using summary statistics available at the GWAS Atlas [13] (https://atlas.ctglab.nl/) from psychiatric, cognitive, neurological phenotype domains, each based on reasonably powered GWAS with sample size of N > 50,000 subjects. Most of these studies are based on UKBB data, and all the studies are based on European ancestry. Since individual-level genotype and phenotype data used in these GWAS are not publicly available, it is very challenging to implement VNTR.g in this context.
Based on a genome-wide significant threshold of p value 5 × 10–8, we did not find any significant association between 5-HTTLPR and the psychiatric phenotypes in PGC (Table 3). For example, the smallest p value was 0.002 for bipolar disorder (Table 3). Among the psychiatric phenotypes analyzed from GWAS atlas, we also did not find any significant association (Table 4 and Supplementary Tables S1 and S2). We provide the GWAS marginal association p values for each pair of the tag SNPs and the psychiatric phenotypes in PGC (Supplementary Table S4).
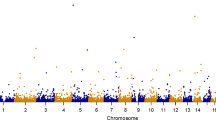
Among the cognitive phenotypes analyzed from this resource, none of the phenotypes was associated; a nearly significant association was observed only for intelligence (Table 5). We also analyzed a set of neurological phenotypes and found no association (Table 5). However, we observed significant associations between 5-HTTLPR and human adult height, BMI, total cholesterol, impedance measures to estimate the trunk fat-free mass, and the trunk predicted mass (Fig. 2). Thus, none of 107 psychiatric phenotypes in GWAS Atlas and nine psychiatric phenotypes in PGC was found to be associated with 5-HTTLPR.

Horizontal bars for the psychiatric phenotypes in the psychiatric genetics consortium (PGC) are colored purple. Rest of these phenotypes were analyzed using summary statistics obtained from studies based on UK Biobank. The vertical dashed bar corresponds to the genome-wide significance threshold –log10(5 × 10–8) = 7.3.
Discussion
We successfully developed and applied a GWAS summary-statistics-based approach to examine the direct effect of the serotonin transporter (SLC6A4) promoter VNTR polymorphism (5-HTTLPR) on susceptibility of neurobehavioral traits and psychiatric disorders in the largest available samples for genetic studies. Despite the long-debated history of this locus as having direct as well as moderating effects on traumatic events and stress on depression [7,8,9,10] (and so many other reports of a wide-range of behavioral phenotypes and personality traits that continue to flood the PubMed publication records), we found no evidence of association between the 5-HTTLPR and any of these traits.
We examined a broad range of psychiatric, cognitive, and neurological phenotypes using publicly available summary statistics. The most significant finding was for association between 5-HTTLPR and intelligence [14] but not reaching the well-established genome-wide significance threshold for common allele associations in GWAS studies [15] and without additional correction for multiple testing given the many traits tested in our study. Further analysis of some 400 disorders and traits in the GWAS Atlas belonging to other domain categories (e.g. environment, immunological, metabolic, respiratory, skeletal) and with reasonably powered GWAS sample sizes of N > 50,000, of which many are based on the UKBB data [13], yielded no significant results except for adult height [13], total cholesterol [16], BMI [17], and two related traits that were available in the UKBB and included in the GWAS Atlas, i.e., impedance measures to estimate the trunk fat-free mass and the trunk predicted mass. Only adult height, however, remains significant after correction for multiple testing. One of the eight SNPs of the 5-HTTLPR predictor, rs11651241, is strongly associated with adult height, which might explain the strong association signal in our study. By and large, our analysis represents the most systematic effort to examine the role of the serotonin transporter promoter polymorphism 5-HTTLPR in neurobehavioral traits without finding evidence for any direct effect of this VNTR to disease or trait susceptibility. Considering our findings and our understanding of the polygenicity of complex traits, together with the challenge of quantitative phenotyping in the realm of neurobehavioral traits and life events, we conclude that the 5-HTTLPR alleles are also very unlikely to play a measurable role in GxE interactions resulting in human disease and behavior.
Minimac3 is a well-known method for genotype imputation in GWAS [18]. It is based on state space reduction of the hidden Markov model, and is computationally much faster than Minimac2 [19]. Border et al. [20] proposed an efficient approach for imputing genotypes of a VNTR polymorphism using Minimac3 and integrating two reference panel datasets containing genotypes of both VNTR variants and genome-wide SNPs, and the haplotype reference consortium data widely used for imputation [21]. They demonstrated good accuracy of the method and applied it to impute VNTR genotypes in the UKBB. When individual-level genotype data of the SNPs surrounding a VNTR locus are available, this approach is useful for imputing the VNTR genotypes. However, due to multiple steps of analysis involved with the method and complex statistical models underlying Minimac3, it is challenging to design the summary-statistics-based version of the approach. Thus, our method is more useful in the absence of individual-level data.
A limitation of our study is that we considered the biallelic short/long alleles of the VNTR polymorphism instead of considering the actual number of repeats. Even though this is a common strategy for association testing, it does not utilize the complete allelic diversity. Since, our approach is adaptable to the prediction of a multi-allelic VNTR variant, it is possible to extend the method to consider the actual number of repeats in the analysis.
Our approach to using GWAS summary statistics for predicting involvement of a VNTR polymorphism in human complex traits, without the need for individual and large-scale PCR-based genotyping, is not limited to the 5-HTTLPR locus. We applied and developed a computational framework that can easily be expanded to include predictors of other types of genetic and genomic variants for testing their involvement in quantitative and qualitative traits and disorders. We showed that the individual-level genotype data-based method and the summary-statistics-based approach (VNTR.g and VNTR.s) perform equally well, which gives confidence about the reliability of the GWAS summary-statistics-based approach. This method is analogous to that of the transcriptome-wide association studies (TWAS), which we previously developed [12] and successfully applied to identify novel genes associated with complex diseases [22,23,24,25]. While the initial TWAS approach is a summary-statistics-based version of predicting gene-expression for association testing, we are now also able to impute (and test for association) the genetic component of allelic variation of more complex genetic and genomic polymorphisms such as VNTRs and other types of recurrent structural variation in the human genome. With the increased efforts of whole genome sequencing of larger cohorts for genetic studies, novel computational methods have been developed to directly test for association of VNTRs, microsatelites, and other repeat structures [26,27,28]. For now, however, with the extensive data collection and availability of much larger GWAS studies with genome genotyping array data of hundreds of thousands of participants, our approach provides a more efficient way of testing for involvement of VNTRs and other types of haplotypes-associated genetic variation in complex traits.
Data availability
The datasets analyzed during the current study are available from the psychiatric genetics consortium (https://www.med.unc.edu/pgc/), UK Biobank (https://www.ukbiobank.ac.uk/), and GWAS atlas (https://atlas.ctglab.nl/). All data generated during this study are included in this article and its supplementary materials.
References
Ripke S, Walters JT, O’Donovan MC; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv [Preprint]. 2020.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JR, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53:817–29.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41.
Singh T, Neale BM, Daly MJ. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. MedRxiv [Preprint]. 2020.
Nakamura Y, Leppert M, O’Connell P, Wolff R, Holm T, Culver M, et al. Variable number of tandem repeat (VNTR) markers for human gene mapping. Science. 1987;235:1616–22.
Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O’Donovan MC, et al. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20:555–62.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–9.
Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–71.
Munafò MR, Durrant C, Lewis G, Flint J. Gene x environment interactions at the serotonin transporter locus. Biol Psychiatry. 2009;65:211–9.
Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. 2011;68:444–54.
Lu AT, Bakker S, Janson E, Cichon S, Cantor RM, Ophoff RA. Prediction of serotonin transporter promoter polymorphism genotypes from single nucleotide polymorphism arrays using machine learning methods. Psychiatr Genet. 2012;22:182–8.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52.
Watanabe K, Stringer S, Frei O, Mirkov MU, de Leeuw C, Polderman TJ, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51:1339–48.
Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, De Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–9.
Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131:747–56.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28:166–74.
Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7.
Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–4.
Border R, Smolen A, Corley RP, Stallings MC, Brown SA, Conger RD, et al. Imputation of behavioral candidate gene repeat variants in 486,551 publicly-available UK Biobank individuals. Eur J Hum Genet. 2019;27:963–9.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat Genet. 2018;50:538–48.
Reus LM, Pasaniuc B, Posthuma D, Boltz T, Ferrari R, Hernandez DG, et al. Gene expression imputation across multiple tissue types provides insight into the genetic architecture of frontotemporal dementia and its clinical subtypes. Biol Psychiatry. 2021;89:825–35.
Feng H, Gusev A, Pasaniuc B, Wu L, Long J, Abu-full Z, et al. Transcriptome-wide association study of breast cancer risk by estrogen-receptor status. Genet Epidemiol. 2020;44:442–68.
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–9.
Dolzhenko E, Van Vugt JJ, Shaw RJ, Bekritsky MA, Van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27:1895–903.
Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt JJ, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020;21:1–4.
Mousavi N, Shleizer-Burko S, Yanicky R, Gymrek M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 2019;47:e90.
Funding
This study was supported by funding R01 MH115676 (BP and RAO) and RF1 AG058484 (RAO).
Author information
Authors and Affiliations
Contributions
Conceptualization: RO and BP. Data curation and formal analysis: PP and AM. Investigation and methodology: AM, PP, BP, and RO. Resources and supervision: RO and BP. Visualization: AM and PP. Writing – original draft: AM, RO, and PP. Writing – review & editing: AM, PP, BP, and RO.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
UCLA IRB approval for the study was provided and it was determined that the research does not involve human subjects as defined in 45 CFR 46.102(f) or 21 CFR 50.3(g).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41431_2021_996_MOESM1_ESM.pdf
Supplementary material: A summary-statistics-based approach to examine the role of serotonin transporter promoter tandem repeat polymorphism in psychiatric phenotype
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Majumdar, A., Patel, P., Pasaniuc, B. et al. A summary-statistics-based approach to examine the role of serotonin transporter promoter tandem repeat polymorphism in psychiatric phenotypes. Eur J Hum Genet 30, 547–554 (2022). https://doi.org/10.1038/s41431-021-00996-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00996-6
This article is cited by
-
2022: the year that was in the European Journal of Human Genetics
European Journal of Human Genetics (2023)
-
Methylation of the serotonin reuptake transporter gene and non-motor symptoms in dystonia patients
Clinical Epigenetics (2022)
-
No gene to predict the future?
European Journal of Human Genetics (2022)
