
Abstract
Decreased or increased activity of potassium channels caused by loss-of-function and gain-of-function (GOF) variants in the corresponding genes, respectively, underlies a broad spectrum of human disorders affecting the central nervous system, heart, kidney, and other organs. While the association of epilepsy and intellectual disability (ID) with variants affecting function in genes encoding potassium channels is well known, GOF missense variants in K+ channel encoding genes in individuals with syndromic developmental disorders have only recently been recognized. These syndromic phenotypes include Zimmermann–Laband and Temple–Baraitser syndromes, caused by dominant variants in KCNH1, FHEIG syndrome due to dominant variants in KCNK4, and the clinical picture associated with dominant variants in KCNN3. Here we review the presentation of these individuals, including five newly reported with variants in KCNH1 and three additional individuals with KCNN3 variants, all variants likely affecting function. There is notable overlap in the phenotypic findings of these syndromes associated with dominant KCNN3, KCNH1, and KCNK4 variants, sharing developmental delay and/or ID, coarse facial features, gingival enlargement, distal digital hypoplasia, and hypertrichosis. We suggest to combine the phenotypes and define a new subgroup of potassium channelopathies caused by increased K+ conductance, referred to as syndromic neurodevelopmental K+ channelopathies due to dominant variants in KCNH1, KCNK4, or KCNN3.
Similar content being viewed by others


Introduction
Potassium (K+) channels form a large and diverse group of ion channels, which are encoded by almost 80 genes in the human genome. K+ channels show diverse gating properties and have various functions in excitable and non-excitable cells. In neurons, K+ channels determine excitability properties and control action potentials in order to maintain excitation homeostasis. K+ channels exhibit broad temporal and spatial expression patterns and regulate cellular excitability during development in multiple ways [1,2,3,4]. Four α subunits arranged around a central pore segment selective for K+ ions are necessary to build a functional potassium channel. According to the number of transmembrane domains, K+ channels are divided into three major groups: the “inward rectifier” K+ (Kir) channel family contains two transmembrane domains, the “two-pore domain leak” K+ (K2P) channel family has four transmembrane domains, and members of the “voltage-gated” K+ (Kv) family, including “calcium-activated” K+ (KCa) channels, have six transmembrane domains. Each α subunit of the channel tetramer has a pore module domain composed of two transmembrane domains, a reentrant pore loop and additional domains which allow responsiveness to different stimuli [4, 5].
Decreased or increased activity of potassium channels caused by loss-of-function and gain-of-function (GOF) variants in the corresponding genes, respectively, underlies a broad spectrum of human disorders affecting the function of the central nervous system, heart, kidney, and other organs [5,6,7,8]. While the association of epilepsy and intellectual disability (ID) with variants affecting function in genes encoding potassium channels has been greatly appreciated [2, 5, 6], GOF missense variants in K+ channel encoding genes in individuals with syndromic developmental disorders have only recently been recognized [9]. One of these syndromic phenotypes is Zimmermann–Laband syndrome (ZLS) (MIM: 135500), a rare disorder characterized by distinct facial dysmorphism with a bulbous nose and thick ears, gingival enlargement, ID with or without epilepsy, hypo- or aplasia of terminal phalanges and nails, and hypertrichosis [10,11,12,13]. Dominant de novo missense variants in KCNH1 (MIM: 603305), encoding the Eag1 (Kv10.1) channel belonging to the ether-à-go-go family of voltage-gated K+ channels, have not only been identified in subjects with ZLS [14], but also in subjects with Temple–Baraitser syndrome (TBS; MIM: 611816) [15]. ZLS and TBS show considerable phenotypic overlap. A total of 23 individuals with KCNH1 variants affecting function have been reported [14,15,16,17,18,19,20]. By studying the biophysical properties of selected KCNH1 mutant channels, a left-shifted current-to-voltage activity and slower deactivation kinetics compared to wild-type channels have been identified, demonstrating a GOF effect for ZLS- and TBS-associated KCNH1 missense variants [14, 15]. In KCNK4 (MIM: 605720), which encodes a two-pore domain leak K+ channel, the two dominant de novo missense variants p.(Ala172Glu) and p.(Ala244Pro) have been reported in three unrelated subjects. They show a consistent phenotype of characteristic facial dysmorphism, hypertrichosis, epilepsy, developmental delay/ID, and gingival overgrowth for which the acronym FHEIG syndrome has been proposed [21]. KCNK4 (alternative names: TRAAK, K2P4.1) belongs to the TRAAK/TREK subfamily of lipid- and mechano-sensitive K2P channels [22]. The two mutant KCNK4 channels showed a higher basal K+ conductance and lacked further channel activation in response to mechanical stimuli and arachidonic acid, indicating a GOF effect of the disease-causing amino acid substitutions [21]. The clinical features of the KCNK4-related disorder are similar to those observed in individuals with ZLS and TBS [14, 15, 21]. In 2019, we reported de novo GOF missense variants in KCNN3 (MIM: 602983) [23], encoding the small-conductance Ca2+-activated K+ channel SK3 that is gated by submicromolar intracellular Ca2+ concentrations [24]. KCNN3/SK3 is a homomeric tetramer and part of a multiprotein complex comprising the pore-forming channel subunits, the constitutively bound Ca2+ sensor calmodulin (CaM), protein kinase CK2 and protein phosphatase 2A (PP2A) [25]. Electrophysiological recordings comparing KCNN3 wild type and p.(Lys269Glu), p.(Gly350Asp), and p.(Ser436Cys) mutants in a heterologous cell system provided evidence for a GOF effect as an increase in Ca2+ sensitivity and a faster activation of the SK3 mutant channels was identified. The three individuals with dominantly acting KCNN3 variants showed moderate developmental delay or mild-to-moderate ID, coarse facial features, gingival enlargement, hypoplasia of distal phalanges, and aplastic or hypoplastic nails; two individuals had patent ductus arteriosus (PDA). These clinical features overlap with the characteristic ZLS phenotype and with the KCNK4-related disorder. For this reason, we propose combining the phenotypes associated with activating KCNH1, KCNK4, and KCNN3 alleles to define a new subgroup of potassium channelopathies caused by increased K+ conductance [23].
Here, we report eight additional individuals with phenotypes belonging to this K+ channelopathy subgroup, including five with KCNH1 missense variants and three with novel KCNN3 variants. We review their clinical features and compare them to 22 previously reported individuals with a KCNH1 GOF allele, the three reported subjects with a dominant KCNK4 variant, and the three individuals previously reported with a dominant KCNN3 variant [14,15,16,17,18,19, 21, 23]. The aim of this study is to better define and differentiate the syndromic phenotypes associated with variants affecting function in KCNH1, KCNK4, and KCNN3 and to delineate the core clinical picture of this subgroup of rare potassium channelopathies.
Subjects and methods
Study approval
Informed consent for genetic analyses was obtained from all patients, and genetic studies were performed clinically. The parents of the affected individuals provided informed consent for participation in the study, clinical data and specimen collection, genetic analysis, and publication of relevant findings. Permission to publish and reproduce previously published photographs was provided for all patients shown in Figs. 1 and 2.

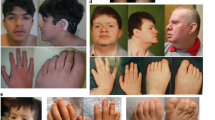
The facial photos are arranged in order of age from youngest to oldest. The five newly reported patients are indicated by P1–P5. Note the hypotonic facial expression, with open mouth posture and inverted V-shape of the upper lip, and apparent ptosis in some individuals. Facial shape elongates with age (third row), but myopathic facial features remain. a, b Patient 3 (P3; at age 16 months) and patient 4 (P4; at age 1 year 7 months) reported in this study (described in detail in Table 1). c Patient at age 3 years reported in [17] (with permission from Springer Nature). d Patient at age 4 years reported in [48] (with permission from John Wiley and Sons). e Patient at age 4 years 4 months reported in [18] (with permission from Springer Nature). f Patient at age 3 years 7 months reported in [49] (with permission from John Wiley and Sons). g Patient at age 6 years reported in [17] (with permission from Springer Nature). h Patient at age 6 years reported in [17] (with permission from Springer Nature). i Patient at age 6 years 10 months reported in [50] (with permission from Wiley and Sons). j Patient at age 7 years reported in [14]. k Patient at age 8 years reported in [17] (with permission from Springer Nature). l Patient at age 9 years reported in [16]. m Patient 2 (P2; at age 9 years) reported in this study (described in detail in Table 1) and previously reported in [18] (individual 3). n Patient at age 12 years reported in [14]. o Patient at age 13 years reported in [18] (with permission from Springer Nature). p Patient at age 12 years 8 months reported in [14]. q Patient at age 14 years reported in [18] (with permission from Springer Nature). r Patient (age unknown) reported in [14]. s Patient 1 (P1; at age 14 years) reported in this study (described in detail in Table 1). t Patient 5 (P5; at age 34 years) reported in this study (described in detail in Table 1). u, v Fingers of patient 3 (P3; at age 16 months; as described in detail in Table 1), showing proximally placed hypoplastic thumbs with hypoplastic nails. w, x Toes of Patient 3 (P3; at age 14 months; as described in detail in Table 1), showing anonychia of toes 1 and 2 and hypoplastic nails on toes 3–5. y, z Toes of patient 4 (P4; at age 3 years 10 months; as described in detail in Table 1), showing elongated toes with hypoplastic nails.

The three newly reported patients are indicated by P6–P8. Note the broad nasal tip, wide mouth, and coarse facial features. Same patient as child (a; at age 5 years) and as adult (b; at age 46 years) after cosmetic facial surgery (previously published in [23]). c, d Two additional individuals, both aged 5 years, previously reported in [23]. e Facial photograph of patient 6 (P6; at age 19 months) reported here (see Table 1 for details) showing epicanthal folds, with distal digital hypoplasia with hypoplastic finger nails (f). g Facial photograph of patient 7 (P7; at age 9 years) reported here (see Table 1 for details), with hypoplastic finger nails (h). i Facial photograph of patient 8 (P8; at age 30 years) reported here (see Table 1 for details), showing full lower lip, with hypoplastic toe nails (j) and hypoplastic finger nails (k).
Exome sequencing (ES) and sequence data analysis
ES for patient 2 has been previously published [18]. For patients 1 and 3–8, ES was performed in clinical diagnostic laboratories (for details, see Supplementary information). Parental samples were included in the analysis as available (Table 1).
Results
Five individuals with a dominant KCNH1 variant and three individuals with a dominant KCNN3 variant
Through an international collaboration, we identified five individuals with a heterozygous KCNH1 missense variant likely affecting function. Clinical and genetic data of the affected individuals are summarized in Table 1. ES revealed the previously described dominant variants p.(Gly496Glu), p.(Arg357Gln), p.(Leu489Phe), and p.(Gly496Arg) in a 39-year-old female (patient 1), a 9-year-old girl (patient 2), a 14-month-old male (patient 3), and a 34-year-old female (patient 5), respectively. Female patient 4, who was 3 years old at last examination, carried the dominant de novo c.1060A > G/p.(Lys354Glu) variant, which has not been reported previously. None of the variants was identified in the gnomAD database. Adult patient 1 had microcephaly, severe ID, absence of speech, and increased tone in limbs. She had a prenatal onset of seizures that persisted after birth. MRI scan at the age of 15 years revealed mild cerebral atrophy and mild cerebellar hypoplasia. Craniofacial dysmorphism comprised narrow face, upslanted palpebral fissures, tented upper lip vermilion, prominent chin, highly arched palate, and gingival enlargement (Fig. 1). She had long and thin hands with adducted thumbs. Her feet were long and narrow with long toes and overriding of 2nd over 3rd toes. Nails were present but had an abnormal shape. She had severe scoliosis. Patient 2 had developmental delay with independent walking at age 3.5 years. She had mixed seizures and ID with no spoken language at age 9 years. Her hypotonic facial features included bitemporal narrowing, and a wide mouth with full lips (Fig. 1). Facial shape appears to elongate with age in these individuals, as seen in patient 5 who as adult had a very long and narrow face with retrognathia (Fig. 1). Her height and head circumference were within the normal range, but she had short distal phalanges and hypoplastic nails. She had moderate ID and epilepsy.
Patients 3 and 4 showed severe developmental delay (DD) and hypotonia. Patient 4 developed seizures at age 4 weeks, while patient 3 did not have seizures. Both had gingival enlargement. The thumbs of patient 3 were proximally placed and hypoplastic. Craniofacial dysmorphism in patient 3 comprised hypotonic and long face, micrognathia and high palate, arched eyebrows, long eyelashes, epicanthal folds, flat nasal bridge, short upturned nose, and tented upper lip (Fig. 1). Patient 4 showed coarse face with epicanthal folds and slightly broad nasal tip (Fig. 1). Patient 3 had anonychia of 1st and 2nd toes and hypoplastic 3rd–5th toes. His thumb nails were hypoplastic (Fig. 1). Patient 4 had elongated toes with hypoplastic nails (Fig. 1) and thoracolumbar scoliosis and kyphosis.
Three individuals with novel dominant KCNN3 variants were recruited: the 1-year-old male patient 6 with the de novo c.1663G > T/p.(Val555Phe) missense change, the 11-year-old female patient 7 with the de novo in-frame deletion c.1616_1618del/p.(Val539del), and the 30-year-old female patient 8 with the c.859G > T/p.(Ala287Ser) missense variant (Table 1). None of the variants was identified in the gnomAD database. Patients 6 and 7 showed mild DD and hypotonia, but no seizures. Patient 8 had mild-to-moderate ID, agenesis of the corpus callosum and mildly increased tone with brisk reflexes, seizures were suspected. Patient 6 had coarse face, synophrys, downslanted palpebral fissures, epicanthal folds, very long eyelashes, low nasal bridge, and broad nasal tip (Fig. 2). Craniofacial dysmorphism in patient 7 comprised thick hair with low anterior and posterior hair lines, thick dark eyelashes, full cheeks with three dimples on the left, depressed nasal bridge and prominent nose with broad tip, full lips, and highly arched palate (Fig. 2). Patient 8 had coarse facial features with a low anterior hairline, long eyelashes, a highly arched palate, and a full lower lip (Fig. 2). Gingival enlargement required surgical reduction in patient 6, was present in patient 7 and absent in patient 8. Distal digital hypoplasia and long great toes were observed in patient 6, while patient 7 only had slight distal shortening of fingers. Patient 8 had broad halluces and the distal phalanges of the toes appeared foreshortened with hypoplastic nails. Patient 6 showed anonychia of thumbs and all toes and extreme nail hypoplasia of 2nd–5th fingers. Patient 7 had small and slowly growing nails. Hypospadias and microcolon were observed in patient 6.
Comparison of clinical features in patients with KCNH1, KCNN3, and KCNK4 variants
We collected clinical information for the eight patients with either a KCNH1 or KCNN3 variant likely affecting function from this study (Table 1), 22 previously reported individuals with a dominant KCNH1 variant [14,15,16,17,18,19], three previously reported subjects with a dominant KCNN3 variant [23], and three previously reported subjects with a dominant KCNK4 missense variant [21]. We did not include the patient with ZLS reported by Guglielmi et al. [20] as the KCNH1 variant was not described and clinical data were sparse. This brings the total number of studied individuals to 36. As in Bramswig et al. [18] (Table 1) and Fukai et al. [17] (Table 1), we focused on the clinical findings suggestive of ZLS, TBS, and/or FHEIG syndromes, such as neurological, skeletal, and nail abnormalities, as well as gingival enlargement and hypertrichosis (Table 2 and Supplementary Tables 1–3). If facial photographs were available, we evaluated craniofacial dysmorphism in the newly reported and published individuals with a variant affecting function in KCNH1, KCNK4, or KCNN3 and defined a facial gestalt associated with dominant variants in either gene (Figs. 1 and 2).
Neurological features
All 36 individuals had DD or ID. Eighteen of 21 (86%) patients with dominant KCNH1 variant had severe DD and the level of ID, determined in 23 individuals, was severe in 22 (96%) and mild to moderate in 1 (4%). In the three individuals with dominant KCNK4 variant, two had severe and one mild-moderate DD and ID. Four subjects with dominant KCNN3 variant had mild or moderate DD and the three oldest individuals, aged 11, 30, and 46 years, had mild ID. Hypotonia was present in 26/27 (96%) individuals with dominant KCNH1 variant, in 4/6 (67%) with dominant KCNN3 variant, and in 2/3 (66%) with dominant KCNK4 variant. The majority of individuals with dominant KCNH1 variant had seizures (24/27; 89%), while only two of the three (66%) with dominant KCNK4 variant developed seizures. Five individuals with dominant KCNN3 variant did not show epilepsy; in one patient seizures were suspected.
Skeletal abnormalities
Finger and toe abnormalities were not reported in the three individuals with dominant KCNK4 variant [21]. All six individuals with dominant KCNN3 variant and 13 of 17 (76%) with dominant KCNH1 variant had hypoplastic terminal phalanges of some or all fingers and/or toes. Broad thumbs and/or toes were present in 11/24 (46%) individuals with dominant KCNH1 variant, while only 1/6 (17%) with dominant KCNN3 variant had these limb abnormalities. Proximal placement and long thumb was observed in 14/18 (78%) cases with dominant KCNH1 and in 1/6 (17%) individuals with dominant KCNN3 variant. Overall, 15/24 (63%) individuals with dominant KCNH1 variant and 2/6 (33%) with dominant KCNN3 variant had long great toes.
Nail anomalies
Absent or hypoplastic thumb nail(s) were observed in 16/27 (59%) individuals with dominant KCNH1 and 5/6 (83%) with dominant KCNN3 variant. In all individuals (100%) with a dominant KCNN3 variant, absence or hypoplasia of great toe nail and of other finger and/or toe nails was present. 24/27 (89%) and 16/20 (80%) patients with dominant KCNH1 variant had absent or hypoplastic great toe nail and anonychia or nail hypoplasia of other fingers and/or toes, respectively. No individual with dominant KCNK4 variant had nail anomaly [21].
Other findings
Gingival enlargement was documented in 15/19 (79%) individuals with dominant KCNH1, in 4/6 (67%) individuals with dominant KCNN3, and in all three (100%) with dominant KCNK4 variant. Similarly, all individuals with dominant KCNK4 variant (100%) had hypertrichosis, while only 3/16 (19%) and 3/6 (50%) with dominant KCNH1 and KCNN3 variant, respectively, showed hypertrichosis.
Craniofacial dysmorphism
We evaluated facial photographs of 20 here reported and previously published individuals with dominant KCNH1 variant and identified the following craniofacial features as most common findings: myopathic and long facies, epicanthal folds, broad nasal tip, and open and wide mouth with tented upper lip vermilion (Fig. 1). The three previously reported and three additional individuals with dominant KCNN3 variant reported here have coarse facial features with thick eyebrows and mild-to-moderate synophrys, prominent nose with a broad nasal tip and triangular nostrils (Fig. 2). Shared craniofacial features in the three individuals with dominant KCNK4 variant comprise bushy and straight eyebrows, long eyelashes, short philtrum, prominent vermillion, and micrognathia [21].
Discussion
We studied a total of 36 individuals with a variant likely affecting function in a potassium channel encoding gene, including eight newly reported individuals, and determined the frequency of overlapping clinical features typical for TBS, ZLS, and FHEIG syndromes in the 27 individuals with dominant KCNH1 variants, six with dominant KCNN3, and three with dominant KCNK4 variants. The data show that the 36 individuals have an overarching clinical picture, however, the phenotypes related to each of the three genes exhibit a distinctive constellation of clinical features that may be recognizable by clinical geneticists. All 36 individuals had DD and/or ID, but of variable degree. While the vast majority of subjects with KCNH1 variant likely affecting function had severe DD and/or severe ID, all patients with KCNN3 variant had a milder form of DD and/or ID. The two individuals with the same KCNK4 variant affecting function [p.(Ala172Glu)] showed severe ID, and they were reported to have nystagmus with bilateral optic hypoplasia. Seizures and/or epilepsy is a typical hallmark of the KCNH1 disorder (89%). None of the six individuals with dominant KCNN3 variant had epilepsy, although seizures could not be excluded in one patient. We evaluated skeletal abnormalities and found finger and toe abnormalities in patients with KCNH1 and KCNN3 variants likely affecting function. Hypoplasia of terminal phalanges is a typical feature in individuals with dominant KCNH1 (76%) and KCNN3 variants (100%). Proximal placement of and long thumb and long great toes were seen in individuals with both dominant KCNH1 (78% and 63%, respectively) and KCNN3 variants (17% and 33%, respectively). Broad thumb and/or toe was observed in 46% of individuals with dominant KCNH1 variant and in 17% with dominant KCNN3 variant. Nail anomalies, such as absence or hypoplasia of finger and/or toe nails, were present in the majority of individuals with dominant KCNH1 (59–89%) and in all with KCNN3 variant. In patients with dominant KCNK4 variant, nail dysplasia was absent, and data on specific finger and toe abnormalities were not reported. The frequency of gingival enlargement and hypertrichosis that are characteristic clinical features of ZLS was determined. Both features were consistently present in individuals with KCNK4 variant affecting function and variably present in patients with dominant KCNN3 (30% for hypertrichosis and 67% for gingival enlargement) or KCNH1 variant (19% for hypertrichosis and 79% for gingival enlargement). Based on the data, the overarching phenotype associated with dominant KCNH1, KCNN3, and KCNK4 variants comprised DD and/or ID, hypotonia, coarsening facial features, gingival enlargement, and hypertrichosis. The greatest clinical overlap was observed between KCNH1- and KCNN3-related disorders as both are additionally characterized by nail and terminal phalangeal aplasia/hypoplasia and additional thumb and toe abnormalities. The degree of DD and/or ID, presence or absence of seizures, broad thumb or toe, and the facial gestalt may help distinguishing between individuals harboring a KCNH1 or KCNN3 variant likely affecting function. On the other hand, the three reported individuals with dominant KCNK4 variant show distinctive facial features with bushy eyebrows, long eyelashes, short philtrum, and prominent vermillion together with consistent gingival enlargement and generalized hypertrichosis that may prompt clinicians to consider FHEIG syndrome.
We propose to define a subgroup of rare potassium channelopathies, which comprises TBS, ZLS, and FHEIG syndromes, all rare developmental and clinically recognizable disorders caused by GOF variants in genes coding for three different membrane-bound potassium channels. Similarly, Hamilton and Suri [9] suggested the term “electrifying dysmorphology” for a group of dysmorphic syndromes characterized by ID, coarse face, gingival overgrowth, hypertrichosis, and digital/toe anomalies that arise from variants in potassium channel encoding genes. Besides ZLS, TBS, and FHEIG syndromes, Hamilton and Suri [9] discussed Birk–Barel syndrome, Andersen–Tawil syndrome, Keppen–Lubinsky syndrome, and Cantú syndrome (CS). We already noticed clinical overlap of ZLS and CS and found early DD, hypertrichosis, gingival enlargement, joint laxity, and hypoplasia of terminal phalanges and nails in one or several of the nine recently reported individuals with a dominant variant in ABCC9 [26]. Dominant variants in ABCC9 and, rarely, in KCNJ8, encoding the regulatory (SUR2) and pore-forming (Kir6.1) subunits, respectively, of ATP-sensitive potassium (KATP) channels cause CS [27,28,29,30]. Distinctive craniofacial features of CS, including coarse facial features, low anterior hairline, wide nasal bridge, epicanthal folds, full lips, and hypertrichosis of the forehead [31], can also be seen in individuals with dominant KCNN3 or KCNK4 variant (Figs. 1 and 2). Typical CS-associated cardiovascular anomalies, including PDA, mild ventricular hypertrophy, hypertrophy of the ventricular septum, and aorta dilatation, may help distinguishing CS from ZLS and/or FHEIG syndrome [26]. Interestingly, PDA, the most frequent cardiac finding in CS (58%) [31], has also been reported in two of six individuals with dominant KCNN3 variant, further showing clinical overlap of CS and the KCNN3-related phenotype.
The biological processes by which pathogenic variants in genes coding for K+ channels lead to complex developmental phenotypes are poorly understood. The KCNH1 encoded Eag1/KV10.1 channel plays a role in cell cycle control and proliferation through cilia disassembly prior to mitosis. Eag1/Kv10.1 hyperactivity due to activating variants has been speculated to cause skeletal and nail malformations through altering signaling pathways involved in morphogenesis, such as the sonic hedgehog pathway [32, 33]. We recently put forward a different hypothesis for the development of digital abnormalities in individuals with KCNN3 variants affecting function [23]. KCNN3 together with KCNN4 channels function in endothelial Ca2+ dynamics to induce vascular tone and blood pressure changes [34,35,36,37]. Local activity of KCNN3 in subspaces of endothelial cells modulates vascular tone and blood pressure [36, 38]. Based on these data we speculated that excessive sustained K+ conductance caused by increased Ca2+ sensitivity of KCNN3 mutant channels leads to enhanced arterial vasodilation and increasing intracapillary pressure during specific stages of embryonic development. This can cause vasodilatory edema, vascular ruptures and/or tissue damage leading to digital hypoplasia or aplasia in individuals with GOF KCNN3 variants. Further evidence for a role of vascular dilation in the development of nail and phalangeal hypoplasia comes from the malformations seen in children with the fetal-hydantoin syndrome [39]. The syndrome is caused by in utero exposure to hydantoin and/or its derivatives such as phenytoin and nifedipine which belong to antiepileptic drugs. These vasodilating drugs cause distal digital defects in rabbits, when given on day 16 of pregnancy that were preceded by edema, hemorrhage and vascular disruption [40, 41]. In neonates with fetal-hydantoin syndrome, hypoplastic finger and toe nails, digitalized great toe and congenital heart diseases, including pulmonary or aortic valvular stenosis, coarctation of aorta and PDA, are characteristic abnormalities [42,43,44]. These clinical features show considerable overlap with CS and ZLS. In addition, administration of the KATP agonist minoxidil causes hypertrichosis as a side effect [45], supporting a link between overactivity of these and other K+ channels and the development of hypertrichosis [46, 47]. Similarly, hypertrichosis and gingival hyperplasia are well recognized side effects of phenytoin use (https://www.aesnet.org/sites/default/files/file). Taken together, studies from teratogens and basic research on potassium channel function suggest that overactivity of these ion channels and increased K+ conductance underlie the clinical similarities seen in a subgroup of potassium channelopathies characterized by DD/ID, epilepsy, coarse facial features, gingival enlargement, hypertrichosis, and/or nail and phalangeal aplasia or hypoplasia.
References
Anderson PA, Greenberg RM. Phylogeny of ion channels: clues to structure and function. Comp Biochem Physiol B Biochem Mol Biol. 2001;129:17–28.
Niday Z, Tzingounis AV. Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist. 2018;24:368–80.
Bauer CK, Schwarz JR. Ether-a-go-go K(+) channels: effective modulators of neuronal excitability. J Physiol. 2018;596:769–83.
Kohling R, Wolfart J. Potassium channels in epilepsy. Cold Spring Harb Perspect Med. 2016;6:a022871.
Allen NM, Weckhuysen S, Gorman K, King MD, Lerche H. Genetic potassium channel-associated epilepsies: Clinical review of the Kv family. Eur J Paediatr Neurol. 2020;24:105–16.
Kessi M, Chen B, Peng J, Tang Y, Olatoutou E, He F, et al. Intellectual disability and potassium channelopathies: a systematic review. Front Genet. 2020;11:614.
Crotti L, Odening KE, Sanguinetti MC. Heritable arrhythmias associated with abnormal function of cardiac potassium channels. Cardiovasc Res. 2020;116:1542–56.
Manis AD, Hodges MR, Staruschenko A, Palygin O. Expression, localization, and functional properties of inwardly rectifying K(+) channels in the kidney. Am J Physiol Ren Physiol. 2020;318:F332–7.
Hamilton MJ, Suri M. “Electrifying dysmorphology”: potassium channelopathies causing dysmorphic syndromes. Adv Genet. 2020;105:137–74.
Castori M, Valiante M, Pascolini G, Leuzzi V, Pizzuti A, Grammatico P. Clinical and genetic study of two patients with Zimmermann-Laband syndrome and literature review. Eur J Med Genet. 2013;56:570–6.
Chacon-Camacho OF, Vazquez J, Zenteno JC. Expanding the phenotype of gingival fibromatosis-mental retardation-hypertrichosis (Zimmermann-Laband) syndrome. Am J Med Genet. 2011;155A:1716–20.
Laband PF, Habib G, Humphreys GS. Hereditary gingival fibromatosis. Report of an affected family with associated splenomegaly and skeletal and soft-tissue abnormalities. Oral Surg Oral Med Oral Pathol. 1964;17:339–51.
Zimmermann KW. Ueber Anomalien des Ektoderms. Vierteljahresschr Zahnheilkd. 1928;44:419–34.
Kortüm F, Caputo V, Bauer CK, Stella L, Ciolfi A, Alawi M, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet. 2015;47:661–7.
Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet. 2015;47:73–7.
Megarbane A, Al-Ali R, Choucair N, Lek M, Wang E, Ladjimi M, et al. Temple-Baraitser syndrome and Zimmermann-Laband syndrome: one clinical entity? BMC Med Genet. 2016;17:42.
Fukai R, Saitsu H, Tsurusaki Y, Sakai Y, Haginoya K, Takahashi K, et al. De novo KCNH1 mutations in four patients with syndromic developmental delay, hypotonia and seizures. J Hum Genet. 2016;61:381–7.
Bramswig NC, Ockeloen CW, Czeschik JC, van Essen AJ, Pfundt R, Smeitink J, et al. ‘Splitting versus lumping’: Temple-Baraitser and Zimmermann-Laband syndromes. Hum Genet. 2015;134:1089–97.
Mastrangelo M, Scheffer IE, Bramswig NC, Nair LD, Myers CT, Dentici ML, et al. Epilepsy in KCNH1-related syndromes. Epileptic Disord. 2016;18:123–36.
Guglielmi F, Staderini E, Iavarone F, Di Tonno L, Gallenzi P. Zimmermann-Laband-1 syndrome: clinical, histological, and proteomic findings of a 3-year-old patient with hereditary gingival fibromatosis. Biomedicines. 2019;7:48.
Bauer CK, Calligari P, Radio FC, Caputo V, Dentici ML, Falah N, et al. Mutations in KCNK4 that affect gating cause a recognizable neurodevelopmental syndrome. Am J Hum Genet. 2018;103:621–30.
Brohawn SG. How ion channels sense mechanical force: insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann N Y Acad Sci. 2015;1352:20–32.
Bauer CK, Schneeberger PE, Kortüm F, Altmuller J, Santos-Simarro F, Baker L, et al. Gain-of-function mutations in KCNN3 encoding the small-conductance Ca(2+)-activated K(+) channel SK3 cause Zimmermann-Laband syndrome. Am J Hum Genet. 2019;104:1139–57.
Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–69.
Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci. 2007;27:2369–76.
Kortüm F, Niceta M, Magliozzi M, Dumic Kubat K, Robertson SP, Moresco A, et al. Cantu syndrome versus Zimmermann-Laband syndrome: report of nine individuals with ABCC9 variants. Eur J Med Genet. 2020;63:103996.
Brownstein CA, Towne MC, Luquette LJ, Harris DJ, Marinakis NS, Meinecke P, et al. Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities—support for the role of K(ATP) channels in this condition. Eur J Med Genet. 2013;56:678–82.
Harakalova M, van Harssel JJ, Terhal PA, van Lieshout S, Duran K, Renkens I, et al. Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat Genet. 2012;44:793–6.
van Bon BW, Gilissen C, Grange DK, Hennekam RC, Kayserili H, Engels H, et al. Cantu syndrome is caused by mutations in ABCC9. Am J Hum Genet. 2012;90:1094–101.
Cooper PE, Reutter H, Woelfle J, Engels H, Grange DK, van Haaften G, et al. Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat. 2014;35:809–13.
Grange DK, Roessler HI, McClenaghan C, Duran K, Shields K, Remedi MS, et al. Cantu syndrome: findings from 74 patients in the International Cantu Syndrome Registry. Am J Med Genet C Semin Med Genet. 2019;181:658–81.
Sanchez A, Urrego D, Pardo LA. Cyclic expression of the voltage-gated potassium channel KV10.1 promotes disassembly of the primary cilium. EMBO Rep. 2016;17:708–23.
Urrego D, Sanchez A, Tomczak AP, Pardo LA. The electric fence to cell-cycle progression: do local changes in membrane potential facilitate disassembly of the primary cilium?: timely and localized expression of a potassium channel may set the conditions that allow retraction of the primary cilium. Bioessays. 2017;39:1600190.
Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–32.
Kohler R, Degenhardt C, Kuhn M, Runkel N, Paul M, Hoyer J. Expression and function of endothelial Ca(2+)-activated K(+) channels in human mesenteric artery: a single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ Res. 2000;87:496–503.
Yap FC, Weber DS, Taylor MS, Townsley MI, Comer BS, Maylie J, et al. Endothelial SK3 channel-associated Ca2+ microdomains modulate blood pressure. Am J Physiol Heart Circ Physiol. 2016;310:H1151–63.
Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–31.
Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601.
Hanson JW. Teratogen update: fetal hydantoin effects. Teratology. 1986;33:349–53.
Danielsson BR, Danielson M, Rundqvist E, Reiland S. Identical phalangeal defects induced by phenytoin and nifedipine suggest fetal hypoxia and vascular disruption behind phenytoin teratogenicity. Teratology. 1992;45:247–58.
Danielsson BR, Danielson M, Reiland S, Rundqvist E, Dencker L, Regard CG. Histological and in vitro studies supporting decreased uteroplacental blood flow as explanation for digital defects after administration of vasodilators. Teratology. 1990;41:185–93.
Hegde A, Kaur A, Sood A, Dhanorkar M, Varma HT, Singh G, et al. Fetal hydantoin syndrome. J Pediatr. 2017;188:304.
Sabry MA, Farag TI. Hand anomalies in fetal-hydantoin syndrome: from nail/phalangeal hypoplasia to unilateral acheiria. Am J Med Genet. 1996;62:410–2.
Silver L. Hand abnormalities in the fetal hydantoin syndrome. J Hand Surg Am. 1981;6:262–5.
Shorter K, Farjo NP, Picksley SM, Randall VA. Human hair follicles contain two forms of ATP-sensitive potassium channels, only one of which is sensitive to minoxidil. FASEB J. 2008;22:1725–36.
McClenaghan C, Huang Y, Yan Z, Harter TM, Halabi CM, Chalk R, et al. Glibenclamide reverses cardiovascular abnormalities of Cantu syndrome driven by KATP channel overactivity. J Clin Invest. 2020;130:1116–21.
Ohko K, Nakajima K, Nakajima H, Hiraki Y, Kubota K, Fukao T, et al. Skin and hair abnormalities of Cantu syndrome: a congenital hypertrichosis due to a genetic alteration mimicking the pharmacological effect of minoxidil. J Dermatol. 2020;47:306–10.
Gabbett MT, Clark RC, McGaughran JM. A second case of severe mental retardation and absent nails of hallux and pollex (Temple-Baraitser syndrome). Am J Med Genet A. 2008;146A:450–2.
Yesil G, Guler S, Yuksel A, Alanay Y. Report of a patient with Temple-Baraitser syndrome. Am J Med Genet A. 2014;164A:848–51.
Jacquinet A, Gerard M, Gabbett MT, Rausin L, Misson JP, Menten B, et al. Temple-Baraitser syndrome: a rare and possibly unrecognized condition. Am J Med Genet A. 2010;152A:2322–6.
Acknowledgements
We thank all patients and family members for their participation in this study. We thank Julie Evans (SW Genomic Laboratory Hub, Bristol Genetics Laboratory, Southmead Hospital, Bristol, UK) for technical advice.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (KO 4576/1-2 to KK). The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (Grant No. HICF-1009-003). This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by Wellcome. See Nature PMID: 25533962 or www.ddduk.org/access.html for full acknowledgment. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KWG is the CMO for FDNA, the company providing the Face2Gene application. IMW and LBH are employees of GeneDx, Inc. HM is an employee and shareholder at Invitae Corporation. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gripp, K.W., Smithson, S.F., Scurr, I.J. et al. Syndromic disorders caused by gain-of-function variants in KCNH1, KCNK4, and KCNN3—a subgroup of K+ channelopathies. Eur J Hum Genet 29, 1384–1395 (2021). https://doi.org/10.1038/s41431-021-00818-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00818-9
This article is cited by
-
Loss of TBC1D2B causes a progressive neurological disorder with gingival overgrowth
European Journal of Human Genetics (2024)
-
Establishment and characterization of ZJUCHi003: an induced pluripotent stem cell line from a patient with Temple–Baraitser/Zimmermann–Laband syndrome carrying KCNH1 c.1070G > A (p.R357Q) variant
Human Cell (2024)
-
Channelopathy of small- and intermediate-conductance Ca2+-activated K+ channels
Acta Pharmacologica Sinica (2023)
-
Intracellular hemin is a potent inhibitor of the voltage-gated potassium channel Kv10.1
Scientific Reports (2022)
-
Potassium Channel KCNH1 Activating Variants Cause Altered Functional and Morphological Ciliogenesis
Molecular Neurobiology (2022)
