
Abstract
Myotonic dystrophy type 1 (DM1) is caused by a CTG trinucleotide repeat expansion on chromosome 19q13.3. While DM1 premutation (36–50 repeats) and protomutation (51–80 repeats) allele carriers are mostly asymptomatic, offspring is at risk of inheriting expanded, symptom-associated, (CTG)n repeats of n > 80. In this study we aimed to evaluate the intergenerational instability of DM1 pre- and protomutation alleles, focussing on the influence of parental gender. One hundred and forty-six parent–child pairs (34 parental premutations, 112 protomutations) were retrospectively selected from the DM1 patient cohort of the Maastricht University Medical Center+. CTG repeat size of parents and children was determined by (triplet-primed) PCR followed by fragment length analysis and Southern blot analysis. Fifty-eight out of eighty-one (71.6%) paternal transmissions led to a (CTG)n repeat of n > 80 in offspring, compared with 15 out of 65 (23.1%) maternal transmissions (p < 0.001). Repeat length instability occurred for paternal (CTG)n repeats of n ≥ 45, while maternal instability did not occur until (CTG)n repeats reached a length of n ≥ 71. Transmission of premutations caused (CTG)n repeats of n > 80 in offspring only when paternally transmitted (two cases), while protomutations caused (CTG)n repeats of n > 80 in offspring in 71 cases, of which 56 (78.9%) were paternally transmitted. In conclusion, our data show that paternally transmitted pre- and protomutations were more unstable than maternally transmitted pre- and protomutations. For genetic counseling, this implies that males with a small DMPK mutation have a higher risk of symptomatic offspring compared with females. Consequently, we suggest addressing sex-dependent factors in genetic counseling of small-sized CTG repeat carriers.
Similar content being viewed by others


Introduction
Myotonic dystrophy type 1 (DM1; OMIM #160900) is an autosomal dominant neuromuscular disorder, caused by a cytosine–thymine–guanine (CTG) repeat expansion on chromosome 19q13.3 [1]. The CTG expansion, located at the 3′untranslated region of the dystrophia myotonica protein kinase (DMPK) gene, seems to alternate RNA-binding protein activity through the production of mutant DMPK transcripts [2]. This pathophysiological process is presumed to give rise to DM1’s clinical features, which vary in age of onset, symptomatology, and severity depending on the DM1 subtype.
From a clinical perspective, DM1 can be divided into four categories, ranging from late-onset to congenital DM1. Late-onset DM1 is associated with repeat lengths <100, congenital DM1 is often associated with repeat lengths >1000, and intermediate phenotypes have repeat lengths in between [3, 4]. While late-onset DM1 may cause early-onset cataract and muscle weakness at older age, congenital DM1 leads to hypotonia and severe respiratory distress at birth [3, 4]. Main features of the remaining two subtypes, known as juvenile and adult-onset DM1, consist of muscular weakness and myotonia in combination with organ involvement, such as cardiac conduction defects or nocturnal hypoventilation [3, 4]. In juvenile and congenital DM1, developmental delay may also be present.
In stable nonpathogenic DMPK alleles, the number of CTG repeats ranges from 5 to 35 per allele [5,6,7,8,9]. Repeat expansions of 36–50 are not associated with symptoms, but are designated DM1 premutations [10, 11]. In case of CTG repeat expansions of 51–150, the diagnosis of DM1 is confirmed if accompanying symptoms are evident [11]. When symptoms of DM1 are absent (asymptomatic family member or fetus), individuals are at risk of developing DM1 [11]. Still, individuals carrying a CTG repeat of 51–80 frequently remain asymptomatic [11,12,13]. These small-sized CTG repeat expansions, that may be transmitted in relatively stable manner for several generations, were designated DM1 protomutations [12]. Repeat expansions > 80, that often cause a strong amplification upon transmission, were designated full-sized DM1 mutations [12, 13].
Genetic anticipation (due to further lengthening of the CTG repeat) is known to cause more severe symptoms, and a decrease in age of onset of DM1 in successive generations [3]. Consequently, it is of great importance to identify CTG repeat expansion carriers, even if these individuals might not be clinically symptomatic. In case of DM1 pre- and protomutation carriers, offspring might develop symptomatic adult-onset, juvenile or even congenital DM1 due to repeat length instability.
Several factors are presumed to play a role in DM1 repeat length stability. While the presence of CTG repeat tract interruptions might work as a stabilizing factor, studies have shown that parental gender can evoke repeat length instability [14,15,16,17,18]. In mothers diagnosed with adult-onset DM1, the CTG repeat seems prone to extreme expansion when transmitted to offspring, resulting in congenital DM1 [17, 18]. For pre- and protomutations, however, a reversed gender effect was suggested. In patients carrying relatively small CTG repeat expansions, paternal transmission caused larger CTG repeat lengths in offspring than maternal transmission [12, 13, 19,20,21].
Still, knowledge of the inheritance of small-sized CTG repeat expansions is scarce, causing uncertainty in genetic counseling. Current guidelines on DNA testing in DM1 describe that small-sized CTG repeats may be unstable and that relatives of carriers are at risk of developing DM1 [11]. However, the effect of parental gender is not addressed.
In this retrospective study, we aim to provide more precise data on gender-dependent intergenerational instability of DM1 pre- and protomutations, in order to improve genetic counseling for DM1 pre- or protomutation allele carriers.
Material and methods
Study population
In order to identify individuals with a pre- or protomutation of de DM1 allele, the DM1 patient cohort database of the Clinical Genetics laboratory of the Maastricht University Medical Center+ was checked for individuals tested between 1993 and 2017. The detection of DM1 pre- or protomutations are mostly the result of family investigation, following the DM1 diagnosis of a symptomatic proband. Family trees were reviewed, to select parent–child pairs in which a pre- or protomutation was transmitted to a successive generation. If a pre- or protomutation carrying parent had multiple children, all children were included as separate parent–child pair. Expanded CTG lengths were categorized based on the mean CTG repeat length (as results are usually given in a range), which was (CTG)n, n = 36–50 in case of a premutation, (CTG)n, n = 51–80 in case of a protomutation, and (CTG)n, n > 80 in case of a full mutation. CTG repeat length instability was defined as an expansion of the pre- of protomutation CTG repeat into (CTG)n, n > 80 in offspring. All CTG repeats used in the study were analyzed as part of regular patient care. Since the determination of DM1 subtypes is based on clinical features, which can become apparent at an older age in case of juvenile, adult- or late-onset DM1, only the occurrence of congenital DM1 in offspring was recorded.
Characterization of CTG repeat lengths
Genomic DNA was extracted from whole blood using the NucleoSpin ®8 Blood Isolation kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions. CTG repeat length analysis was performed by CTG-PCR, followed by Southern blot hybridization, and triplet-primed PCR (TP-PCR). For the CTG-PCR, 200 ng genomic DNA was used with 20 pmol Fw-P1 primer 5′-AGAAAGAAATGGTCTGTGATCCC-3′, 20 pmol 6-FAM labeled Rv-P2 primer 5′-GAAGGGTCCTTGTAGCCGGGAA-3′, and 10% DMSO. TP-PCR was performed on both strands of the CTG repeat according to Warner et al. [22]. After PCR amplification, the fragments were analyzed on ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Subsequently, Southern blot analysis was performed to confirm the CTG-PCR and TP-PCR results. Therefore, PCR products obtained using Fw-P1 and Rv-P2 were separated on a 1.5% agarose gel and blotted onto a Nylon membrane (Roche Diagnostics GMbH, Mannheim, Germany). The membrane was hybridized with a 5′digoxygenine labeled (CTG)10 oligo probe. After stringency washes, the digoxygenine label was visualized using anti-digoxigenin-AP-conjugate and CDP-Star (Roche Diagnostics GMbH, Mannheim, Germany).
CTG repeat interruptions were determined by modification of the TP-PCR. The Rv-P2 primer was replaced by the Fw-P1 primer, and P4CAG primer (tacgcatcccagtttgagacgCAGCAGCAGCAGCAG) by a GGC or CCG specific primer (tacgcatcccagtttgagacgTGCCGCTGCCGCTGCC and tacgcatcccagtttgagacgTGGGCCTGGGCCTGGGC, respectively). After PCR amplification, the fragments were analyzed on ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). CTG tract interruptions in DM1 pre- and protomutations were confirmed by DNA sequencing. All collected repeat sequences were submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/; accession numbers SCV001156422–SCV001156483).
Statistical analysis
Statistical analysis was performed using IBM SPSS statistics software version 24 (SPSS Inc, Chicago, IL, USA). The distribution of continuous variables was assessed for normality by visual inspection of histograms and standardized normal probability plots. Continuous variables are expressed as mean ± standard deviation when normally distributed. Categorical variables are expressed as counts with corresponding percentages. Qualitative data were compared using the chi-squared (χ2) test or Fisher’s exact test, quantitative data were compared using the unpaired Student’s t test. P < 0.05 was considered statistically significant.
Results
Patient characteristics
A total of 146 parent–child pairs with a known parental pre- ((CTG)n, n = 36–50) or protomutation ((CTG)n, n = 51–80) of the DM1 locus were included. Thirty-four (23.3%) parents carried a premutation and 112 (76.7%) carried a protomutation (Fig. 1). A total of 35 parents with multiple children were included. Of the transmitting parents, 81 were male and 65 were female. Baseline characteristics of included parents are further summarized in Table 1.

Individuals with an identified premutation ((CTG)n, n = 36–50) or protomutation ((CTG)n, n = 51–80), tested between 1993 and 2017, were selected from the myotonic dystrophy type 1 (DM1) patient cohort database of the Clinical Genetics laboratory of the Maastricht University Medical Center+. Intergenerational instability of the CTG repeat length is displayed for both groups separately. Out of 146 pre- and protomutation transmissions, 97 transmissions regard first-born offspring.
Intergenerational instability
Upon transmission in the premutation group, 2 out of 34 alleles (5.9%) expanded into (CTG)n of n > 80 in offspring. In the protomutation group, 71 out of 112 alleles (63.4%) expanded into (CTG)n of n > 80 in offspring (Fig. 1). Chi-square analysis indicated a significant association between the type of mutation (pre- or protomutation) and expansion of the CTG repeat into (CTG)n, n > 80 (p < 0.001). None of the children that inherited a (CTG)n of n > 80 were affected by the congenital subtype of DM1. CTG tract interruptions were observed in two DM1 premutation carrying parents and their offspring.
Intergenerational instability in relation to parental gender
The combined results of both pre- and protomutation transmissions show that 58 out of 81 (71.6%) paternal transmissions lead to a (CTG)n repeat of n > 80 in offspring, in comparison with 15 out of 65 (23.1%) maternal transmissions (p < 0.001) (Table 1). Repeat length instability occurred for the entire range of paternal (CTG)n repeats of n ≥ 45, while maternal instability was not observed until (CTG)n repeats reached a length of n ≥ 71 (Fig. 2). Apart from the observed CTG thresholds in our study population ((CTG)n, n = 45 for paternal transmission and (CTG)n, n = 71 for maternal transmission), paternal transmission of DM1 pre- and protomutations seemed to be more frequently unstable than maternal transmission, even if the (CTG)n repeat in offspring did not reach lengths over n > 80 (Fig. 3).

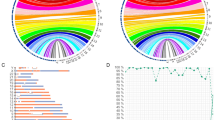
The diagrams demonstrate the relationship between parental CTG repeat length and DM1 status in offspring, for DM1 premutation ((CTG)n, n = 36–50) and protomutation ((CTG)n, n = 51–80) allele carrying parents. DM1 status in offspring is based on a cut-off value of (CTG)n, n > 80. a Paternal transmission. b Maternal transmission.

Charts have been categorized by parental gender. DM1 status is based on a cut-off value of (CTG)n, n > 80. The values above each pie chart represent parental CTG repeat length.
When restricting the analysis to first-born offspring (n = 97), paternal transmission of pre- and protomutations lead to (CTG)n of n > 80 in offspring in 41 out of 57 cases (71.9%). For maternal inheritance, transmission of pre- and protomutations lead to (CTG)n of n > 80 in offspring in 10 out of 40 cases (25%). Chi-square analysis indicated a significant association between parental gender and expansion of the CTG repeat into (CTG)n, n > 80 in case of first-born offspring (p < 0.001). The observed sex-dependent CTG threshold of (CTG)n, n ≥ 45 for males and (CTG)n, n ≥ 71 for females remained the same.
Transmission of premutations (n = 34) caused (CTG)n repeats of n > 80 in offspring in two cases, of which both were paternally transmitted. Transmission of protomutations (n = 112) caused (CTG)n repeats of n > 80 in offspring in 71 cases, of which 56 (78.9%) were paternally transmitted.
Inheritance pattern in case of multiple children
For 35 DM1 pre- or protomutation allele carrying parents, we had information about the CTG repeat size in more than one child. In 31 of these families, all tested children were in the same CTG category (either all tested children had a (CTG)n of n > 80, or all tested children carried a pre- or protomutation). In four of the investigated families, some of the tested children inherited a (CTG)n repeat of n > 80, while others inherited a relatively stable form of the pre- or protomutation. In two of these families, only the second child inherited a (CTG)n of n > 80 (one out of two children, 50%). In a family of three children, the second and third child had a (CTG)n of n > 80 (66.7%). In another family of five children, only the third child (20%) was affected by DM1 with (CTG)n, n > 80. In these four families, all transmitting parents were of male gender.
Discussion
In this study we retrospectively evaluated the intergenerational instability of DM1 pre- and protomutation alleles, focussing on the influence of parental gender. Our results show that paternal transmission of both pre- and protomutations is relatively unstable, causing (CTG)n repeats of n > 80 in offspring in 71.6% of cases. For maternal transmission, only 23.1% of offspring inherited a (CTG)n repeat of n > 80. Moreover, the (CTG)n threshold for DM1 pre- and protomutation instability in our study population was n ≥ 45 for fathers, in comparison with (CTG)n, n ≥ 71 for mothers. In general, DM1 protomutations were far less stable than DM1 premutations.
Some studies have previously reported on the influence of parental gender on DM1 pre- and protomutation transmission [12, 13, 17]. These studies support that paternal transmission is more likely to cause repeat length instability than maternal transmission [12, 13, 17]. In these studies, however, the proportions of paternal unstable transmission were even higher than the 71.6% found in our study [12, 17]. Possibly, this was caused by an overestimation due to smaller sample sizes and broader inclusion ranges (parental (CTG)n repeats up to n = 100).
We also report on gender-dependent CTG thresholds for the inheritance of small-sized repeat expansions (Fig. 2). Paternal repeat length instability started from (CTG)n repeats as low as n = 45. Although the CTG threshold for female inheritance is based on only 15 cases, the higher threshold value further validates the role of parental sex. Moreover, paternal transmission was found to be of a more unstable nature, even if CTG repeats in offspring did not exceed (CTG)n, n = 80 (Fig. 3).
None of the children in our study population were affected by the congenital subtype of DM1. The congenital subtype is typically the result of maternal inheritance of adult DM1 with (CTG)n repeats of n > 150 [11, 17, 18, 23]. Thus, it may seem as if maternally transmitted large CTG repeat expansions are more unstable than paternally transmitted repeat expansions. This apparent reversed relationship between parental gender and CTG repeat size in offspring might be explained by negative selection of large DM1 alleles at spermatogenesis [17, 24, 25]. Consequently, paternally transmitted small-sized CTG repeats would demonstrate substantial lengthening of the repeat in offspring, as observed in our study, while paternally transmitted congenital DM1 would be rare due to natural selection. This proposed mechanism is strengthened by the limited number of case reports on paternally inherited congenital DM1, in which paternal repeat lengths were mostly on the lower end of the scale ((CTG)n between n = 65 and n = 200) [25,26,27,28,29]. In our study population, however, large repeat expansions in offspring and congenital DM1 were not observed, which could be considered a limitation that possibly attenuates the aforementioned hypothesis.
In other trinucleotide repeat disorders, such as Huntington disease (HD), parental gender differences in transmission have also been observed [30,31,32,33]. For HD, large CAG expansions in offspring occur almost exclusively through paternal transmission, while maternal transmission was found to be relatively stable [32]. HD intermediate alleles, which resemble DM1 pre- and protomutations since they rarely cause clinical symptoms but are prone to intergenerational instability, show a similar effect of paternal sex [31].
Still, parental gender does not seem to be the only factor contributing to DM1 pre- or protomutation instability. As our results show, different children of the same transmitting parent can inherit either (CTG)n repeats of n > 80, or a relatively stable form of the pre- or protomutation. This phenomenon was observed in four families, in which the transmitting parent was the father. The length of the parental repeat expansions itself seems to be of influence on allele instability (Fig. 3), but the literature suggests that other factors such as parental age, genetic modifiers, and DNA repair mechanisms might also play a role [12, 25, 33, 34]. Since first-born offspring in these four families did not inherit (CTG)n repeat lengths of n > 80, paternal somatic instability of the repeat might have also had an effect.
Apart from this, it is known that repeat stability can be influenced by DMPK allele methylation and by the presence of interruptions in the DMPK CTG repeat tract [35,36,37,38]. DMPK allele methylation was not assessed in this observational study, since determination of the methylation profile is not part of routine DMPK repeat length analysis. While the exact role of CTG repeat interruptions has not been clarified, they seem to be present in <5% of DM1 patients and can possibly cause stability or even contraction of the CTG repeat [35, 36, 39]. In our study population, CTG tract interruptions were demonstrated in only two DM1 premutation carrying parents and their offspring. Thus, the effect of CTG tract interruptions in this study seems limited.
For the interpretations of our results, it is important to consider the role of DM1 tissue heterogeneity. Previous research has shown that somatic variation of CTG repeat expansions can contribute to observed intergenerational differences of CTG repeat lengths [19, 40]. Moreover, for larger repeats, the size of the determined CTG repeat expansion appears to be age-dependent and test results could therefore differ after several years [40, 41]. However, somatic variation seems to be less prominent and independent of age for small-sized DM1 alleles, presumably minimizing its effect [13, 19].
In the current study, several parent–child pairs with the same transmitting parent were included. In order to determine the potential influence of multiple transmissions, we repeated our analysis while restricting it to first-born offspring. This analysis determined that the percentages of DM1 affected offspring in first-born children were comparable with the results for the entire group of 146 DM1 parent–child pairs.
There are, however, some limitations to this study. First of all, data were collected in a retrospective manner and it should be noted that new mechanistic assays were not performed. Moreover, pre- or protomutation carriers were identified through clinically affected probands. Families in which the pre- or protomutation has been transmitted in a relatively stable manner over several generations, without the presence of a clear clinical picture, are therefore missed in the study. This could have resulted in an overestimation of intergenerational instability. Collecting prospective data on pre- and protomutation transmissions in families of the general population, without knowledge of an affected proband, seems unachievable however. Yet, the current study describes the largest cohort of parent–child pairs in which a small-sized CTG repeat was transmitted to the next generation. In addition, the results of both pre- and protomutation transmissions were combined in a single study, describing possible sex-dependent CTG thresholds for the first time.
In clinical practice, the results of this study can be of significant value when counseling small-sized CTG repeat allele carriers. Guidelines for molecular testing in DM1 describe that small-sized CTG repeats ((CTG)n, n = 36–50) may be unstable, but are non-specific and ignore the role of parental gender [11]. We recommended to counsel pre- or protomutation allele carriers in a specific manner, addressing the influence of parental gender. In case of a male small-sized repeat carrier, the risk of having a symptomatically affected child is considerably large, as paternal instability was observed to start for (CTG)n, n = 45. For female allele carriers, the risk is lower, but increases as the maternal CTG repeat lengthens. Based on these risk figures, the choice of future parents to opt for natural pregnancy with or without prenatal testing, or preimplantation genetic testing may be individualized.
In conclusion, we found that paternal transmission of DM1 pre- and protomutations is far more unstable compared with maternal transmission of small-sized CTG repeats. We also found a lower CTG thresholds for the instability of paternal DM1 pre- and protomutation alleles. Ultimately, we recommend a sex-dependent genetic counseling advice for DM1 small-sized repeat carriers.
References
Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;69:385.
Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–55.
Harper PS. Myotonic dystrophy present management, future therapy. Oxford: Oxford University Press; 2004.
Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochimica et Biophysica Acta. 2015;1852:594–606.
Imbert G, Kretz C, Johnson K, Mandel JL. Origin of the expansion mutation in myotonic dystrophy. Nat Genet. 1993;4:72–6.
Rubinsztein DC, Leggo J, Amos W, Barton DE, Ferguson-Smith MA. Myotonic dystrophy CTG repeats and the associated insertion/deletion polymorphism in human and primate populations. Hum Mol Genet. 1994;3:2031–5.
Goldman A, Ramsay M, Jenkins T. Absence of myotonic dystrophy in southern African Negroids is associated with a significantly lower number of CTG trinucleotide repeats. J Med Genet. 1994;31:37.
Neville CE, Mahadevan MS, Barcelo JM, Korneluk RG. High resolution genetic analysis suggests one ancestral predisposing haplotype for the origin of the myotonic dystrophy mutation. Hum Mol Genet. 1994;3:45–51.
Zerylnick C, Torroni A, Sherman SL, Warren ST. Normal variation at the myotonic dystrophy locus in global human populations. Am J Hum Genet. 1995;56:123–30.
Yamagata H, Miki T, Sakoda S, Yamanaka N, Davies J, Shelbourne P, et al. Detection of a premutation in Japanese myotonic dystrophy. Hum Mol Genet. 1994;3:819–20.
Kamsteeg EJ, Kress W, Catalli C, Hertz JM, Witsch-Baumgartner M, Buckley MF, et al. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet. 2012;20:1203–8.
Barcelo JM, Mahadevan MS, Tsilfidis C, MacKenzie AE, Korneluk RG. Intergenerational stability of the myotonic dystrophy protomutation. Hum Mol Genet. 1993;2:705–9.
Martorell L, Monckton DG, Sanchez A, Lopez de Munain A, Baiget M. Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology. 2001;56:328–35.
Ashizawa T, Dubel JR, Dunne PW, Dunne CJ, PhD YHF, Md AP, et al. Anticipation in myotonic dystrophy: II. Complex relationships between clinical findings and structure of the GCT repeat. Neurology. 1992;42:1877–83.
Hunter A, Tsilfidis C, Mettler G, Jacob P, Mahadevan M, Surh L, et al. The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J Med Genet. 1992;29:774.
Shelbourne P, Winqvist R, Kunert E, Davies J, Leisti J, Thiele H, et al. Unstable DNA may be responsible for the incomplete penetrance of the myotonic dystrophy phenotype. Hum Mol Genet. 1992;1:467–73.
Brunner HG, Brüggenwirth HT, Nillesen W, Jansen G, Hamel BC, Hoppe RL, et al. Influence of sex of the transmitting parent as well as of parental allele size on the CTG expansion in myotonic dystrophy (DM). Am J Hum Genet. 1993;53:1016–23.
Redman JB, Fenwick RG Jr., Fu YH, Pizzuti A, Caskey CT. Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. J Am Med Assoc. 1993;269:1960–5.
Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8.
Abbruzzese C, Costanzi Porrini S, Mariani B, Gould FK, McAbney JP, Monckton DG, et al. Instability of a premutation allele in homozygous patients with myotonic dystrophy type 1. Ann Neurol. 2002;52:435–41.
Pratte A, Prevost C, Puymirat J, Mathieu J. Anticipation in myotonic dystrophy type 1 parents with small CTG expansions. Am J Med Genet A. 2015;167a:708–14.
Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, et al. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–6.
Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet. 1993;52:1164–74.
Jansen G, Willems P, Coerwinkel M, Nillesen W, Smeets H, Vits L, et al. Gonosomal mosaicism in myotonic dystrophy patients: involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm. Am J Hum Genet. 1994;54:575–85.
de Die-Smulders CE, Smeets HJ, Loots W, Anten HB, Mirandolle JF, Geraedts JP, et al. Paternal transmission of congenital myotonic dystrophy. J Med Genet. 1997;34:930–3.
Zeesman S, Carson N, Whelan DT. Paternal transmission of the congenital form of myotonic dystrophy type 1: a new case and review of the literature. Am J Med Genet. 2002;107:222–6.
Bergoffen J, Kant J, Sladky J, McDonald-McGinn D, Zackai EH, Fischbeck KH. Paternal transmission of congenital myotonic dystrophy. J Med Genet. 1994;31:518–20.
Nakagawa M, Yamada H, Higuchi I, Kaminishi Y, Miki T, Johnson K, et al. A case of paternally inherited congenital myotonic dystrophy. J Med Genet. 1994;31:397–400.
Ohya K, Tachi N, Chiba S, Sato T, Kon S, Kikuchi K, et al. Congenital myotonic dystrophy transmitted from an asymptomatic father with a DM-specific gene. Neurology. 1994;44:1958–60.
Semaka A, Creighton S, Warby S, Hayden MR. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin Genet. 2006;70:283–94.
Semaka A, Collins JA, Hayden MR. Unstable familial transmissions of Huntington disease alleles with 27-35 CAG repeats (intermediate alleles). Am J Med Genet B Neuropsychiatr Genet. 2010;153b:314–20.
Kremer B, Almqvist E, Theilmann J, Spence N, Telenius H, Goldberg YP, et al. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet. 1995;57:343–50.
Losekoot M, van Belzen MJ, Seneca S, Bauer P, Stenhouse SAR, Barton DE, et al. EMQN/CMGS best practice guidelines for the molecular genetic testing of Huntington disease. Eur J Hum Genet. 2013;21:480–6.
Neto JL, Lee JM, Afridi A, Gillis T, Guide JR, Dempsey S, et al. Genetic contributors to intergenerational CAG repeat instability in Huntington’s disease knock-in mice. Genetics. 2017;205:503–16.
Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A. 2009;149a:1365–74.
Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19:1399–412.
Barbé L, Lanni S, López-Castel A, Franck S, Spits C, Keymolen K, et al. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am J Hum Genet. 2017;100:488–505.
López Castel A, Nakamori M, Tomé S, Chitayat D, Gourdon G, Thornton CA, et al. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet. 2010;20:1–15.
Pesovic J, Peric S, Brkusanin M, Brajuskovic G, Rakocevic-Stojanovic V, Savic-Pavicevic D. Molecular genetic and clinical characterization of myotonic dystrophy type 1 patients carrying variant repeats within DMPK expansions. Neurogenetics. 2017;18:207–18.
Wong LJ, Ashizawa T, Monckton DG, Caskey CT, Richards CS. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am J Hum Genet. 1995;56:114–22.
Martorell L, Monckton DG, Gamez J, Johnson KJ, Gich I, Lopez de Munain A, et al. Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients. Hum Mol Genet. 1998;7:307–12.
Acknowledgements
We would like to thank Linda Meekels and Chantal Calis for their analytical support, and Stacha Reumers for data collection support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Joosten, I.B.T., Hellebrekers, D.M.E.I., de Greef, B.T.A. et al. Parental repeat length instability in myotonic dystrophy type 1 pre- and protomutations. Eur J Hum Genet 28, 956–962 (2020). https://doi.org/10.1038/s41431-020-0601-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0601-4
