
Abstract
Consanguinity, commonplace in many regions around the globe, is associated with an increased risk of autosomal recessive (AR) genetic disorders. Consequently, consanguineous couples undergoing preimplantation genetic diagnosis (PGD) for one Mendelian disorder may be at increased risk for a child with a second, unrelated AR genetic disorder. We examined the yield of exome analysis for carrier screening of additional AR disorders, beyond the primary diagnosis, amongst consanguineous vs. non-consanguineous populations. Parental samples from trio exomes of 102 consanguineous families and 105 non-consanguineous controls were evaluated for shared carrier status, after disregarding the primary molecular diagnosis. Results were sub-classified according to disease severity. Secondary shared carrier status for pathogenic and likely pathogenic variants leading to AR disorders of moderate to profound severity was identified in 10/102 (9.8%) consanguineous couples, as compared to 1/105 (0.95%) non-consanguineous couples (χ2 = 8.0565, p value < 0.005). Higher inbreeding coefficient values, calculated from individual exomes, correlated with secondary shared carrier status for diseases of moderate to profound severity (r = 0.17, p value < 0.0125). Our results indicate that consanguineous couples undergoing PGD are at increased risk for a second genetic disease of moderate to profound severity. This study represents an underestimate of the rate of secondary shared carrier status due to inability to detect deep intronic variants, no assessment of copy number variants, and false negative results stemming from stringent variant interpretation. False positive results may result from inaccuracies in public databases. Additional studies in consanguineous populations will determine whether exome-based carrier screening should be recommended to all couples undergoing PGD.
Similar content being viewed by others

Introduction
A significant portion of the world population lives in communities with a strong preference for consanguineous marriage [1, 2]. Such marriages are traditional in most communities of North Africa, the Middle East and West Asia, accounting for 20–50% of all marriages. In addition, consanguineous marriages are frequent among emigrant communities now resident in Europe, North America, or Australia [1]. While consanguineous marriages are important in keeping social stability and enforcing family solidarity, they also have medical consequences, in particular increasing the risk for congenital malformations and genetic diseases [2,3,4,5]. The additive risk is mainly attributed to shared carrier status for homozygous variants associated with autosomal recessive (AR) genetic disorders [1].
The main goal of exome/genome sequencing in the clinical setting is to reach a primary molecular diagnosis for a monogenic disorder given an affected proband. Yet, an important byproduct of these analyses is the ability to detect medically actionable secondary findings [6, 7]. Moreover, identification of shared parental carrier status in the case of trio exomes (i.e., when both parents have a variant in the same gene causing a disease with AR inheritance) is important for future family planning [8,9,10].
The concept of multilocus variation leading to multiple Mendelian diagnoses, both within individuals and within families, has been well documented [11,12,13]. We hypothesized that consanguineous couples are more likely to have shared carrier status for a second AR diagnosis, unrelated to the primary diagnosis in their affected child. This would place consanguineous couples undergoing preimplantation genetic diagnosis (PGD) at risk of having a child who is unaffected by the primary disease addressed by the PGD, but affected by a second, unrelated disorder. This would also challenge the common practice of pursuing proband-only exomes and not trio exomes in closely consanguineous families [5], especially in light of the decreasing costs of exomes. In order to address these questions, we undertook a retrospective, systematic analysis of exome-based shared carrier status of pathogenic and likely pathogenic variants, in consanguineous vs. non-consanguineous couples.
Materials and methods
Study design
We conducted a retrospective analysis in couples who underwent exome sequencing as part of a trio exome. Between the years 2012–2019, the vast majority of exomes undertaken at our center in consanguineous families was by a proband-only approach, and only 102 exomes were analyzed by a trio approach. The main consideration was financial, and most trios were done during the latter part of the study (2018–2019), as prices of exomes decreased. The degree of relation between spouses was determined during genetic counseling. Self-reported consanguinity (first cousins, first cousins once removed, and second cousins) was corroborated with an inbreeding coefficient (F) ≥ 3.125% in the offspring’s exome. 105 non-consanguineous couples, representing the 105 most recent trios generated the same time period (regardless of ancestry), served as controls.
Exome sequencing analysis
Following informed consent, exonic sequences were enriched from genomic DNA with the SureSelect Human All Exon 50 Mb V5/ 60 Mb V6 Kit (Agilent Technologies, Santa Clara, California, USA). Sequences were generated on a HiSeq2500 (Illumina, San Diego, California, USA) as 125-bp paired-end runs or NovaSeq 6000 as 150-bp paired-ends runs. Read alignment and variant calling were performed with DNAnexus (Palo Alto, California, USA) using default parameters, with the human genome assembly hg19 (GRCh37) as reference. Variants were then filtered out if >8 bp from splice junction, synonymous (unless 0–3 nucleotides from an exon-intron boundary), or were seen over 20 times in the homozygous state in the GnomAD database (https://gnomad.broadinstitute.org/).
Calculation of the inbreeding coefficient, F
Estimation of the inbreeding coefficient (F) was based on detection of runs of homozygosity (ROH) in individual exomes. FROH for an individual exome was calculated using the “DetectRUNS” package in R as:
where \(\mathop {\sum }\nolimits^ L_{ROH}\) is the sum of length of all the ROHs detected in the exome, and \(L_{genome}\) is the total length of the genome used (see Table S1 for specific parameters).
Detection of shared carrier status
An in-house automated python-based script was devised to screen parental exomes for shared carrier status of clinical significance, as well as X-linked variants (which served as an internal control). Reported variants included either known published assumed-pathogenic variants in public databases (ClinVar, Human Gene Mutation Database (HGMD), or the Israeli National Genetic Database (INGD)) or loss-of-function variants in known disease-associated genes: stop-gain, frameshift, and splice donor/acceptor variants (Fig. S1). The script is available upon request. Each final document was manually curated in order to remove false positive variants (i.e., conflicting interpretations on ClinVar with benign classifications overriding pathogenic classifications or, otherwise, with insufficient evidence for pathogenicity; homozygous loss-of-function (HLOF) variants in genes known to be tolerant to such variants; genes with well-described pseudogenes affecting alignment, etc.). In addition, each variant was classified according to the ACMG classification [7] by at least two independent physicians/bioinformaticians. The terms “pathogenic” and “likely pathogenic” in this manuscript refer to variants which are disease-causative or likely disease-causative when biallelic or when inherited in trans to a second pathogenic or likely pathogenic variant. Discrepancies in variant classification were resolved by discussion. Variants were submitted to the ClinVar database (NCBI).
Classification of allele severity
Severity of a particular variant was determined based on the classification suggested by Lazarin et al., into one of four groups: profound, severe, moderate, or mild [14]. Diseases with available treatments (i.e., biotinidase deficiency, glucocorticoid deficiency) were classified based upon the severity of the untreated disease. If a particular variant was previously published or deposited into a public database (i.e., ClinVar, HGMD), the severity was classified based on the knowledge accumulated with regards to that specific variant.
Results
Case reports
Overall, three major categories of trio exome results could be defined (Fig. S2). In the first class, the proband had an unequivocal primary molecular diagnosis and parents shared carrier status for a secondary, unrelated diagnosis. Notably, the second AR diagnosis could not be attributed to physical linkage of the two disease loci. The second category included cases where the proband did not have a straightforward primary molecular diagnosis, yet parents were found to be carriers for a second diagnosis unrelated to the phenotype of the proband. In some families, a sibling had already been born with the second diagnosis and Sanger sequencing confirmed homozygosity of the relevant variant in that sibling. In the third category of cases, neither a definitive primary nor secondary diagnosis could be identified. Several exemplary case reports are provided in the Supplemental Data.
Secondary findings in consanguineous vs. non-consanguineous couples
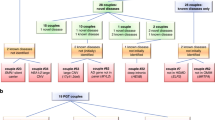
In order to assess the yield of exome-based carrier screening for secondary diagnoses in parents of affected probands, we conducted a retrospective analysis in consanguineous vs. non-consanguineous couples. In the consanguineous group, the distribution according to ancestry included 97 Arab couples (95.1%) and 5 Jewish couples (4.9%) (Fig. 1a, Table S2). The non-consanguineous group included 78 Jewish couples of various origins (74.3%), 26 Arab Muslim couples (24.8%), and one couple of Christian European origin (Fig. 1a, Table S3). Thus, our cohort represented the local population and the social norms, where close consanguinity is most prevalent among Arab Muslims [15].

a Distribution of consanguineous and non-consanguineous couples according to ancestry. b Distribution of secondary shared findings among consanguineous and non-consanguineous couples based on disease severity.
We next set to determine the shared carrier status for a second diagnosis. Notably, shared carrier status for the primary diagnosis associated with the referral indication was disregarded from further analyses, due to the inherent ascertainment bias. Interestingly, in some trios (as in cases of Family 33 and Family 55, supplemental data), the proband did not have a definitive primary molecular diagnosis, yet parents were found to share a carrier status for at least one disorder unrelated to the referral diagnosis (i.e., the proband in such cases was either wild-type or heterozygous for the parental shared variant). Overall, shared carrier status for AR disorders was identified in 15/102 (14.7%) of consanguineous couples after disregarding the primary diagnosis (Table 1). Among the secondary shared variants, at least 4 were private variants, not documented previously in in-house or publically available databases. Two couples had two secondary shared variants (GUCY2C and NADK2 in Family 12 and DHCR7 and ATP7B in Family 33). All couples in this group carried an identical variant in the respective genes; none had compound heterozygous carrier status. By comparison, only 7/105 (6.7%) of the non-consanguineous couples shared autosomal carrier status in the same gene. Among these, 2 couples carried identical variants and 5 couples had different variants (i.e., offspring had 25% risk of being compound heterozygous). In order to control for unexpected biases in the genetic constitution of the two groups, we analyzed and compared maternal X-linked carrier status in both groups. The consanguineous and the non-consanguineous groups had an equivalent number of cases (five) with a pathogenic G6PD variant.
Consanguineous couples have a high rate of shared variants in genes associated with diseases of moderate to profound severity
In order to assess the clinical implications of the observed secondary findings, these were separated into four categories which relied upon a classification of disease severity previously suggested by Lazarin et al. [14]. Briefly, moderate to profound genetic disorders are those that justify preventive measures. Based on this classification, 10/102 (9.8%) consanguineous couples or 10 of 15 (66.7%) consanguineous couples with secondary findings carried shared variants in genes causing disease of moderate, severe or profound severity. Notably, two couples had two shared variants: the first had shared variants in both NADK2 (severe phenotype) and in GUCY2C (mild phenotype) and the other had shared variants in both DHCR7 (profound) and ATP7B (severe). 5/102 (4.9%) carried shared variants only in genes causing diseases of mild severity. In the non-consanguineous group, a single couple out of 105 (0.95%) were compound carriers of likely pathogenic variants in a gene that causes disease of moderate severity. The other 6/105 (5.7%) had variants in genes causing phenotypes of mild severity (Fig. 1b).
When comparing the two groups, there were more secondary shared findings in the consanguineous group as compared to the non-consanguineous group (χ2 = 3.5204, p value < 0.061). The difference was more pronounced and achieved statistical significance when genes associated with diseases of moderate to profound (but not mild) severity were compared (χ2 = 8.0565, p value < 0.0046). Moreover, the point-biserial correlation coefficient between the coefficient of inbreeding (F) and secondary shared carrier status for diseases of moderate to profound severity was r = 0.17 (p value < 0.0125), indicating a positive correlation. As one would expect, secondary compound shared variants were observed only in the non-consanguineous group. This observation could not be attributed to ascertainment bias and approached statistical significance (two tailed p value < 0.06 using Fisher’s exact test).
Discussion
Consanguinity increases the prevalence of AR disorders through inheritance of pathogenic family-specific or population-specific variants [13]. Early awareness of shared variants is important for family planning, as it can be addressed by PGD [16, 17]. The main question driving this study was whether the increased risk of having other children with an independent AR disorder would justify recommending exome sequencing of parents to all couples planning to undergo PGD for a specific Mendelian condition. This was sparked by clinical observations of such families (see Supplemental Data) and paucity of systematic analyses addressing this. Although expanded carrier screening tests are widely available nowadays, the advantage of extracting such data from exome sequencing is the ability to identify variants outside specific panels [8, 9, 18, 19]. This becomes critical when dealing with very rare or yet undetected family-specific variants, as is often the case within consanguineous families [13, 20, 21].
We identified an increased prevalence of shared carrier status for a secondary AR disease of moderate to profound severity in the consanguineous group (9.8%) versus the non-consanguineous group (0.95%). Shared carrier status for genes associated with moderate to profound phenotypes showed a positive correlation with the coefficient of inbreeding r = 0.17 (p value < 0.0125). The absolute number of shared carrier status for a secondary AR disease of mild severity was identical in both groups, possibly due to the higher minor allele frequency (MAF) of variants causing mild disease as opposed to severe disease. Additionally, shared carrier status in X-linked genes was equal amongst the two groups, suggesting that although the groups were not matched by ancestry, this did not result in a significant bias. Our findings are consistent with that of Monies et al., who demonstrated that among 503 couples of the highly inbred Saudi population, over 12% shared at least one common pathogenic variant, beyond the main causative variant relating to the referral diagnosis [13].
The statistics presented herein represent an underestimation of the overall shared carrier rate for secondary AR diseases, due to several limitations. First, if a proband had two AR diagnoses [22], both were disregarded since both were considered related to the referral diagnosis. If we were to consider one diagnosis as a primary diagnosis and the accompanying diagnosis as a secondary diagnosis, this would introduce an ascertainment bias. Second, missense variants of unknown significance, even if affecting a highly conserved residue and predicted pathogenic, were not included in this analysis. Variant interpretation is particularly challenging when considering shared carrier status, since ideally these are identified before an affected child is born and correlation with a phenotype is not an option. Stringent variant interpretation is inevitable in order to avoid misinterpretation of benign variants, and parents must be aware of this limitation of shared carrier screening. We believe that stringent variant interpretation resulted in more false negative results than false positive results, but undoubtedly, false positive results may also be encountered due to misclassification of variants in public databases as pathogenic or likely pathogenic [23, 24]. Finally, pathogenic copy number variants (CNVs, deletions and duplications), well documented in AR disorders [25, 26], were beyond the scope of this study and would be expected to further increase the percentage of shared carrier status. Other technical limitations of exome sequencing in preconceptional screening include inability to detect deep intronic variants, and challenges in interpretation of CNVs in genes with pseudogenes such as SMN1 associated with spinal muscular atrophy [MIM 253300] (albeit, this may be assessed with relative confidence) [27] and CYP21A2 associated with congenital adrenal hyperplasia [MIM 210910].
Potential biases of the study include the fact that controls were not ancestry-matched. All populations examined in this study have genetic carrier screening programs available for recessive diseases with a carrier frequency of 1:60 or greater, along with a recommendation to consider screening for less frequent diseases; however, the compliance with performing these and the number of diseases addressed in each population may create a bias. A second potential bias is in the severity classification scheme. Two of the AR diseases are treatable in a manner that significantly alters the natural history of the severe/profound disease (Table 1), and PGD in such cases should be weighed against the possibility of life-long treatment and surveillance. Layperson perceptions of risk and impact of genetic conditions and of disease severity may differ from expert perception, and should be considered when counseling families on carrier screening [28].
In conclusion, results from the present study suggest that in many cases undergoing PGD, both spouses of consanguineous couples may be at risk for a severe genetic disease which is not the primary reason for the procedure. This knowledge is of utmost importance to the couple and the medical team in order to plan for the future. Genetic counseling of the family should include an explanation regarding the challenges of variant interpretation and the risk of false negative results due to variants of unknown significance [21], so as to minimize undue anger if an affected child is born despite trio exome sequencing and PGD. Couples with one severe condition and a second mild or potentially treatable condition may wish to prioritize transfer of embryos unaffected by both diseases followed by embryos with only a mild or treatable disease, in order to avoid futile PGD cycles. We urge for additional studies in consanguineous populations in order to determine whether our results can be replicated in other populations and, if yes, whether to recommend parental exome-based carrier screening before PGD in such couples.
Data availability
The ClinVar accession numbers for the DNA variant data are SCV001430581.1-SCV001430595.1 and SCV001437665-SCV001437673.
References
Hamamy H, Antonarakis SE, Cavalli-Sforza LL, Temtamy S, Romeo G, Kate LP, et al. Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med. 2011;13:841–7.
Becker R, Keller T, Wegner RD, Neitzel H, Stumm M, Knoll U, et al. Consanguinity and pregnancy outcomes in a multi-ethnic, metropolitan European population. Prenat Diagn. 2015;35:81–9.
Shieh JT, Bittles AH, Hudgins L. Consanguinity and the risk of congenital heart disease. Am J Med Genet A. 2012;158A:1236–41.
Bennett RL, Motulsky AG, Bittles A, Hudgins L, Uhrich S, Doyle DL, et al. Genetic counseling and screening of consanguineous couples and their offspring: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2002;11:97–119.
Eaton A, Hartley T, Kernohan KD, Ito Y, Lamont RE, Parboosingh JS, et al. When to think outside the autozygome: Best practices for exome sequencing in “consanguineous” families. Clin Genet. 2020;97:835–43.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Capalbo A, Valero RA, Jimenez-Almazan J, Pardo PM, Fabiani M, Jiménez D, et al. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: translational research genomic data from 14,125 exomes. PLoS Genet. 2019;15:e1008409.
Gambin T, Jhangiani SN, Below JE, Campbell IM, Wiszniewski W, Muzny DM, et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. 2015;7:54.
Sallevelt SCEH, de Koning B, Szklarczyk R, Paulussen ADC, de Die-Smulders CEM, Smeets HJM. A comprehensive strategy for exome-based preconception carrier screening. Genet Med. 2017;19:583–92.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coabn Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl J Med. 2017;376:21–31.
Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med. 2018;20:1528–37.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;105:879.
Lazarin GA, Hawthorne F, Collins NS, Platt EA, Evans EA, Haque IS. Systematic classification of disease severity for evaluation of expanded carrier screening panels. PLoS One. 2014;9:e114391.
Sharkia R, Mahajnah M, Athamny E, Khatib M, Sheikh-Muhammad A, Zalan A. Changes in marriage patterns among the Arab community in Israel over a 60-year period. J Biosoc Sci. 2016;48:283–7.
Delatycki MB, Alkuraya F, Archibald A, Castellani C, Cornel M, Grody WW, et al. International perspectives on the implementation of reproductive carrier screening. Prenat Diagn. 2020;40:301–10.
Zlotogora J, Grotto I, Kaliner E, Gamzu R. The Israeli national population program of genetic carrier screening for reproductive purposes. Genet Med. 2016;18:203–6.
Antonarakis SE. Carrier screening for recessive disorders. Nat Rev Genet. 2019;20:549–61.
Guo MH, Gregg AR. Estimating yields of prenatal carrier screening and implications for design of expanded carrier screening panels. Genet Med. 2019;21:1940–7.
Josephi-Taylor S, Barlow-Stewart K, Selvanathan A, Roscioli T, Bittles A, Meiser B, et al. User acceptability of whole exome reproductive carrier testing for consanguineous couples in Australia. J Genet Couns. 2019;28:240–50.
Kirk EP, Barlow-Stewart K, Selvanathan A, Josephi-Taylor S, Worgan L, Rajagopalan S, et al. Beyond the panel: preconception screening in consanguineous couples using the TruSight One “clinical exome”. Genet Med. 2019;21:608–12.
Berkun L, Slae M, Mor-Shaked H, Koplewitz B, Eventov-Friedman S, Harel T. Homozygous variants in MAPRE2 and CDON in individual with skin folds, growth delay, retinal coloboma, and pyloric stenosis. Am J Med Genet A. 2019;179:2454–8.
Shah N, Hou YC, Yu HC, Sainger R, Caskey CT, Venter JC, et al. Identification of misclassified ClinVar variants via disease population prevalence. Am J Hum Genet. 2018;102:609–19.
Xiang J, Yang J, Chen L, Chen Q, Yang H, Sun C, et al. Reinterpretation of common pathogenic variants in ClinVar revealed a high proportion of downgrades. Sci Rep. 2020;10:331.
Boone PM, Campbell IM, Baggett BC, Soens ZT, Rao MM, Hixson PM, et al. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome Res. 2013;23:1383–94.
Yuan B, Wang L, Liu P, Shaw C, Dai H, Cooper L, et al. CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genet Med. 2020;22:1633–41.
Liu B, Lu Y, Wu B, Yang L, Liu R, Wang H, et al. Survival motor neuron gene copy number analysis by exome sequencing: assisting spinal muscular atrophy diagnosis and carrier screening. J Mol Diagn. 2020;22:619–28.
Leo MC, McMullen C, Wilfond BS, Lynch FL, Reiss JA, Gilmore MJ, et al. Patients’ ratings of genetic conditions validate a taxonomy to simplify decisions about preconception carrier screening via genome sequencing. Am J Med Genet A. 2016;170:574–82.
Acknowledgements
The authors wish to thank Prof. Joël Zlotogora for thought-provoking discussions and critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mor-Shaked, H., Rips, J., Gershon Naamat, S. et al. Parental exome analysis identifies shared carrier status for a second recessive disorder in couples with an affected child. Eur J Hum Genet 29, 455–462 (2021). https://doi.org/10.1038/s41431-020-00756-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00756-y
Keywords
This article is cited by
-
2021 at European Journal of Human Genetics: the year in review
European Journal of Human Genetics (2022)
