
Abstract
In 1994, a kindred from Yemen was described as the first Jewish family with Machado–Joseph disease (MJD/SCA3), a dominant ataxia caused by the expansion of a (CAG)n above 61 repeats, in ATXN3. MJD is spread worldwide due to an ancient variant of Asian origin (the Joseph lineage). A second, more recent, independent expansion arose in a distinct haplotype (Machado lineage); other possible origins are still under study. We haplotyped 46 MJD patients and relatives, from 6 Israeli Yemenite families, and 100 normal chromosomes from that population, for 30 SNPs spreading 15 kb around the (CAG)n, and 8 STRs and 1 indel in the flanking regions. All six families shared an extended haplotype, showing no variants or recombination after a common origin, but differing in two SNPs (rs12895357 and rs12588287) from the Joseph lineage. To test for a new mutational origin in this population, we searched for the presence of that haplotype in Yemenite-Jewish controls. Only one (1%) normal (CAG)32 allele showed an extended STR-haplotype genetically closer to MJD than normal haplotypes (genetic distance, DA, 0.43 versus 0.53). That normal allele could be explained either by (1) the introduction of both normal and expanded alleles carrying this “Joseph-like” haplotype into the genetic pool of the Yemenite population; or by (2) a large contraction from the expanded CAG range. Based on the lack of STR diversity in MJD Yemenite-Jewish families, and on high frequency of this Joseph-like haplotype among African controls (23.2%), expanded alleles seem to have been introduced very recently (<400 years ago) from Africa.
Similar content being viewed by others

Introduction
Machado–Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), is the most frequent dominant ataxia worldwide, but its prevalence varies significantly among populations, its highest relative frequency, among all spinocerebellar ataxias, being reported from China (62.1%) [1], Brazil (59.6%) [2], Portugal (57.8%) [3], Thailand (46.5%) [4], Germany (42%) [5] and Singapore (41%) [6] (reviewed in [7]). In Israel, in spite of its ethnically diverse population, MJD has been exclusively reported among Jewish families of Yemenite origin [8, 9].
Jews arrived in Yemen mostly in the second century, maintaining close communal structures. By the end of the nineteenth century, Yemenite Jews started to migrate to Israel, where about 350,000 of their descendants now live. In 1994, Goldberg-Stern et al. described the first Israeli Jewish family with a clinical description of MJD, originating from a remote village near Ta’izz in Yemen [8]; the molecular diagnosis of MJD was confirmed in 1996 [9]. Recently, disease prevalence was estimated to be as high as 29:100,000 in Jews of Yemenite descent living in Israel [10].
Due to the large pleomorphism of MJD, three sub-phenotypes were defined [11]: type 1 with an earlier age-at-onset (AO; mean, 24.6 years) and characterised by striking pyramidal and extrapyramidal signs; type 2 (mean AO, 40.3 years) with an intermediate severity, and dominated by progressive ataxia, pyramidal signs and progressive external ophthalmoplegia (PEO); and type 3 with a later onset (mean AO, 47.1 years) and progressing slowly with peripheral signs, in addition to PEO and cerebellar and pyramidal signs. A type 4 was later suggested, including neuropathy and Parkinsonism, and was mainly observed in African patients [12, 13]. These sub-phenotypes do overlap and onset is classically as a type 2 in virtually all cases [14]. In Yemenite Jews, despite the existing variability, type 3 is the most common, whereas type 1 is rare [10].
The variant responsible for MJD is a CAG repeat within an exonic region of ataxin-3 (ATXN3; 14q32.12; NM_001164778.1(ATXN3):c.458CAG[2]CAA[1]AAG[1]CAG[1]CAA[1]CAG[8]), usually expanded above 61 units in patients; normal alleles typically range 12–44 CAGs [15,16,17,18]. To study the ancestral origins of MJD, we have previously assessed SNP backgrounds in patients from 264 MJD families, from 20 populations [19]. Mutation rate of SNPs is very low (~2 × 10–8), the reason why mutations giving rise to most SNPs are considered unique events during the evolution of a given species. We identified two stable SNP haplotypes in MJD, TTACAC and GTGGCA, named “Joseph” and “Machado” lineages, after their predominance in Flores and São Miguel (the Azorean islands home to the Joseph and Machado families, respectively). A Joseph-like (“Groote”) lineage is present in Australian aborigine and some Asian MJD families [20].
Taking into account that MJD has not been observed in other Jewish Israeli subpopulations, nor in other ethnic groups living or originating in Yemen, we aimed at assessing the mutational origin of MJD families of Yemenite-Jewish descent.
Subjects and methods
Subjects
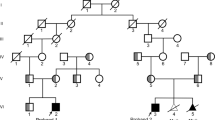
We studied MJD patients (n = 27) and relatives (n = 19), from six Yemenite-Jewish families living in Israel, who emigrated from different villages in Yemen and showed no consanguinity. A total of 100 normal chromosomes were analysed from 30 healthy individuals from the same population and 12 non-affected family members, together with 16 non-expanded chromosomes carried by patients. This study has been approved by the Meir Medical Centre Ethics Committee. All participants gave written consent, after being informed about the research purpose. DNA samples were labelled with a numeric code at the Meir Medical Centre, before being sent to i3S for genotyping (with relevant partial pedigrees).
Genotyping
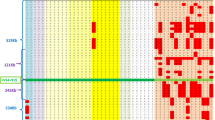
We used a haplotyping approach, as already described [21]. We genotyped SNPs in MJD patients and controls by sequencing a 4 kb region flanking the ATXN3-(CAG)n and a more distant fragment (~12 kb from the repeat), where four additional SNPs have been previously studied in MJD families (Fig. 1); genotyping was performed as described before [20]. Genotyping of STRs was carried out in a single multiplex PCR reaction, optimised to amplify all eight markers. Reactions were done in a final volume of 20 µL, with 5 µL of Taq PCR Master Mix Kit Qiagen® (QIAGEN, Hilden, Germany), 1.5 µL of Q-Solution (QIAGEN, Hilden, Germany) and 15 ng/µL DNA. Concentration of primers was 0.25 µM (AAAC_123 (NC_000014.9:g.91948295GTTT[7]), AC_21 (NC_000014.9:g.92050144GT[13]), GT_199 (NC_000014.9:g.92269835AC[25]), GT_190 (NC_000014.9:g.91881121AC[17]), TG_191 (NC_000014.9:g. 92261998CA[19])) or 0.125 µM (TAT_223 (NC_000014.9:g.92294367ATA[14]), ATA_194 (NC_000014.9:g.92265563TAT[11]) and AC_190 (NC_000014.9:g.91880527GT[18])). The genotyping of rs67740495 (NC_000014.9:g.92262055_92262056del) was done by sequencing, together with STR TG_191, due to their close physical distance. Initial denaturation was performed at 95 °C for 15 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 62 °C for 90 s and extension at 72 °C for 60 s; and final extension at 70 °C for 30 min.

Location of SNPs, Indel and STRs flanking the ATXN3_CAG repeat analysed in this study. Distances (kb) from the (CAG)n are included in the name of analysed STRs; others are in parentheses. Asterisks show SNPs previously studied (haplotypes TTACAC and GTGGCA for Joseph and Machado lineages, respectively).
Analyses
Haplotypes were inferred by segregation, in MJD families, and by segregation combined with the use of PHASE v2.1.1 [22], in controls. Phylogenetic networks were performed using Network 5.0.0.1 [23] and POPTREE2 [24]. Since we used microsatellite data, a combined reduced median and median-joining calculation was done to reduce reticulation. We drew phylogenetic networks by using seven molecular markers (TG_191 was not included due to its complexity); weight for each STR was calculated, based on molecular diversity, with Arlequin 3.5.2.2 [25]. The most recent common ancestor was estimated as previously described [19]. To calculate genetic distance (DA) between STR haplotypes of JC6 and the remaining controls and MJD haplotypes, we used the neighbour-joining method of phylogenetic reconstruction.
To estimate the age for the introduction of MJD expanded alleles in this Jewish community, we relied on known STR mutation rate (μ) and recombination rate (c) between STRs and MJD expansions as a molecular clock. Thus, the probability of change in ancestral haplotypes per generation is ε = 1−[(1−c)(1−μ)]. Taking into account that average of mutation and recombination events on the ancestral haplotype is given by
the number of generations, t, elapsed since a common ancestral, can be calculated as λ=εt.
Results
A new SNP background is shared by all Yemenite-Jewish MJD families
We identified 30 SNPs flanking the ATXN3-(CAG)n, 22 of which distinguish the main (Machado and Joseph) MJD lineages. All six Yemenite-Jewish families with MJD shared a single SNP-based haplotype, which differed from the Joseph lineage in only two SNPs: rs12895357 and rs12588287 (Table 1). To assess the possibility of a de novo expansion having occurred among Jews of Yemenite descent, we constructed SNP-based haplotypes in 100 non-expanded chromosomes from this population. A single-normal chromosome carrying the same haplotype as patients was found in one of the controls (1%), a non-affected mother carrying alleles (CAG)23 and (CAG)32. Taking into account that rs12895357 is located immediately next to the (CAG)n (1 bp), recombination is unlikely; however, if the downstream haplotype with the two variants of rs12895357 and rs12588287 were very common in controls, recombination would become more plausible. Thus, we analysed data from the 1000 Genomes Project (http://www.internationalgenome.org/) to assess frequency of the potentially recombinant downstream haplotype in the major ethnic control groups: 23.2% (307/1322) in African populations, 2.6% (18/694) in mixed American populations, 0% (0/1006) in Europe and 2.97% (59/1986) in Asia.
STR diversity within the newly identified MJD background
A shared extended haplotype, including eight STRs and one indel, was observed in all six families: 16–25–10-del-22-(CAG)exp-14–7–19–25. Taking into account the high mutation rate of STRs, this shows that the Yemenite-Jewish MJD families must share a recent ancestor. This haplotype is phylogenetically close to the one found in one control chromosome (JC6, which carries the same SNP background as the MJD families; Fig. 2a): 16–22–10-del-24-(CAG)32–16–7–19–24. This could reinforce the hypothesis of a de novo expansion having occurred on this background, even if frequency of the new MJD background was as low as 1% among controls from a matched Yemenite-Jewish population. To clarify this question, we calculated genetic distances and performed phylogenetic reconstruction between the JC6 control, other Yemenite-Jewish controls, and MJD families (Fig. 2b). The haplotype of the control JC6 is genetically closer to MJD haplotypes than to other normal control haplotypes (genetic distance, DA, 0.43 versus 0.53). Thus, (1) this normal JC6 chromosome has been recently introduced in the gene pool of Yemenite-Jews (namely, from the African population, where frequency of this Joseph-like background is as high as 23.2%), or (2) this rare normal haplotype has arisen by a large contraction of an expanded MJD allele. Larger normal repeats were rare in controls, this (CAG)32 being the only allele over 30 CAGs (Table 2), what may strengthen the second alternative. Also, most STR alleles flanking this (CAG)32 were not the most frequent when we looked at the control population (except for AC_190 and GT_190). As for the indel marker, the insertion allele is the most frequent worldwide, with the exception of Eastern Asian populations (data from Ensemble; rs67740495).

Phylogenetic networks showing the most parsimonious relationships among STR-haplotypes flanking the ATXN3-(CAG)n in six Yemenite-Jewish families with MJD, 100 Yemenite-Jewish control chromosomes, and the single-control haplotype (JC6) found to share the same Joseph-like SNP background (Joseph lineage with variants in rs12895357 and rs12588287) as MJD Israeli patients. a Circle size is proportional to number of chromosomes/families; nodes represent haplotypes not found in our populations. b Phylogenetic reconstruction based on genetic distances (DA) between the three groups analysed
Age estimation for the presence of MJD among Yemenite Jews
These Yemenite-Jewish families with MJD must share a very recent ancestor, given the lack of STR diversity. To estimate the maximum time of their divergence, we simulated a scenario where a variant in one of the analysed STRs had been detected in one of the six families, i.e. an average for mutation/recombination events (λ) of 1/6. We also calculated probability of change per generation (Ɛ), considering both mutation (µ) and recombination (c) rates, as previously described [19]. The physical distance between the two farthest STRs analysed is 413.9 kb; using a conversion factor of 1.41 cM for each Mb, the recombination fraction for these STRs (0.58 cM apart) would equal 0.0058. Mutation rate for trinucleotides (6.13 × 10−4) was calculated as the median between the rate for di (7.8 × 10−4) and tetranucleotides (4.46 × 10−4). As we typed 5 di, 2 tri and 1 tetranucleotide markers, the average mutation rate was estimated at 6.96 × 10−4; taking into account the eight STRs studied, µ would equal eight times this value, i.e., 5.57 × 10−3.
Given that λ = Ɛt, where t is the number of generations, and that Ɛ = 1-[(1-c)(1-µ)], and assuming a generation time of 25 years, then 0.167 = 1.134 × 10−2t; i.e. an estimated 368 years must have ensued from a common ancestor for all six MJD families.
Discussion
The Joseph and Machado MJD lineages differ in such a large number of SNPs (both up and downstream the CAG expansion) that a scenario of (at least) two independent mutational origins is very likely for MJD. More complex SNP data may, however, be difficult to interpret. An SNP background observed to segregate with expanded alleles could be (1) the signature of a de novo expansion that occurred on this background, but also (2) a complex scenario resulting from recombination and/or recurrence of SNPs on a pre-existing disease background. Clarification of this is of great importance, not only from an epidemiological point of view (e.g. geographical differences in disease prevalence could be explained by de novo variants and diverse frequency of risk haplotypes, or due to general population genetics factors as migration, founder effects or other); but also to study basic mechanisms of (CAG)n instability underlying instability and de novo expansions. Here, we analysed affected families and a matched control population, and supplemented SNP genotyping with analysis of flanking STRs, after phasing both SNP and STR variants segregating with the MJD expansion.
The finding of MJD among Yemenite Jews led to question whether its presence in this isolated Jewish community was due to a new mutational event. All affected families showed a new SNP background, not previously associated to MJD; however, the fact that only two SNPs (rs12895357 and rs12588287) differed from the previously identified Joseph lineage led us to pursue alternative scenarios to explain it. If recombination were to explain this new (Joseph-like) MJD haplotype, it should have happened among Africans or people of African descent who later migrated to the Middle East. Alternatively, two recurrent SNP mutations would have occurred on the Joseph lineage. Previously, strong evidence pointed to the occurrence of a recurrent mutation at one of these SNP (rs12895357) on the Machado lineage (GTGGCA), explaining the GTGCCA haplotype found in three Azorean MJD families [19]. Thus, this may be an atypical SNP, with a higher than average mutation rate. In the Jewish families, however, a back mutation G > A in rs12588287 must have occurred on the same background. If so, the change reverted the allele to its ancestral state, the more frequent A allele (MAFderived allele G = 0.25). Under that scenario, it is highly unlikely that the two SNP reverse mutations arose simultaneously on the Joseph background; hence, we would expect to find expanded haplotypes with just one or the other variant, what was not observed. Interestingly, rs12895357 is one of the three SNPs analysed in a worldwide study, in which three MJD families (two from the United States and one from Morocco) differed only by a G (instead of C) at rs12895357 from the Joseph haplotype [26]. It is unknown, however, whether these three families also share the same variant in rs12588287 as the Jewish patients. If they do not, then they would carry a putative (intermediate) haplotype linking the Joseph and this Joseph-like Yemenite-Jewish haplotype. On the other hand, if they all shared the two variants, this Joseph-like lineage would not be exclusive of Jewish families, but would be older and may have spread from Africa or the Middle East.
To test this hypothesis, we assessed a more distant region flanking up and downstream the (CAG)n, by genotyping fast-evolving STRs. All MJD families analysed shared a single-extended haplotype (including eight STRs and one indel), showing that the Yemenite-Jewish MJD families must have a (very) recent common ancestor. Therefore, in case the new SNP haplotype in Jewish families is observed in other populations, its place-of-birth is unlikely to be in the Middle East.
Based on the diversity accumulated due to STR mutation or recombination, we estimated at 368 years the maximum time elapsed since a common ancestor for all MJD Yemenite-Jewish families, most likely to have been introduced from Africa. This is in contrast with what we have previously found in other populations, where MJD origins were much older [19]. To discern whether this common ancestor is the result of a new mutational origin for MJD or of the introduction of this Joseph-like haplotype in Yemenite Jews, we will now extend this more comprehensive haplotype study to other MJD populations worldwide.
References
Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, et al. Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. J Cent South Univ Med Sci. 2011;36:482–9.
de Castilhos RM, Furtado GV, Gheno TC, Schaeffer P, Russo A, Barsottini O, et al. Spinocerebellar ataxias in Brazil--frequencies and modulating effects of related genes. Cerebellum. 2014;13:17–28.
Vale J, Bugalho P, Silveira I, Sequeiros J, Guimaraes J, Coutinho P. Autosomal dominant cerebellar ataxia: frequency analysis and clinical characterization of 45 families from Portugal. Eur J Neurol. 2010;17:124–8.
Boonkongchuen P, Pongpakdee S, Jindahra P, Papsing C, Peerapatmongkol P, Wetchaphanphesat S, et al. Clinical analysis of adult-onset spinocerebellar ataxias in Thailand. BMC Neurol. 2014;14:75.
Schols L, Amoiridis G, Buttner T, Przuntek H, Epplen JT, Riess O. Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol. 1997;42:924–32.
Zhao Y, Tan EK, Law HY, Yoon CS, Wong MC, Ng I. Prevalence and ethnic differences of autosomal-dominant cerebellar ataxia in Singapore. Clin Genet. 2002;62:478–81.
Martins S, Sequeiros J. Origins and spread of Machado–Joseph disease ancestral mutational events. In: Almeida LP, Nóbrega C, editors. Adv Exp Med Biol. 2018;1049:243–54.
Goldberg-Stern H, D’Jaldetti R, Melamed E, Gadoth N. Machado–Joseph (Azorean) disease in a Yemenite Jewish family in Israel. Neurology. 1994;44:1298–301.
Lerer I, Merims D, Abeliovich D, Zlotogora J, Gadoth N. Machado–Joseph disease: correlation between the clinical features, the CAG repeat length and homozygosity for the mutation. Eur J Hum Genet. 1996;4:3–7.
Zaltzman R, Sharony R, Klein C, Gordon CR. Spinocerebellar ataxia type 3 in Israel: phenotype and genotype of a Jew Yemenite subpopulation. J Neurol. 2016;263:2207–14.
Coutinho P, Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology. 1978;28:703–9.
Subramony SH, Hernandez D, Adam A, Smith-Jefferson S, Hussey J, Gwinn-Hardy K, et al. Ethnic differences in the expression of neurodegenerative disease: Machado–Joseph disease in Africans and Caucasians. Mov disord. 2002;17:1068–71.
Gwinn-Hardy K, Singleton A, O’Suilleabhain P, Boss M, Nicholl D, Adam A, et al. Spinocerebellar ataxia type 3 phenotypically resembling parkinson disease in a black family. JAMA Neurol. 2001;58:296–9.
Coutinho P. Doença de Machado-Joseph: Tentativa de definição. University of Porto, Portugal; 1992.
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–8.
Maciel P, Gaspar C, DeStefano AL, Silveira I, Coutinho P, Radvany J, et al. Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am J Hum Genet. 1995;57:54–61.
Maciel P, Costa MC, Ferro A, Rousseau M, Santos CS, Gaspar C, et al. Improvement in the molecular diagnosis of Machado-Joseph disease. JAMA Neurol. 2001;58:1821–7.
Maruyama H, Nakamura S, Matsuyama Z, Sakai T, Doyu M, Sobue G, et al. Molecular features of the CAG repeats and clinical manifestation of Machado-Joseph disease. Hum Mol Genet. 1995;4:807–12.
Martins S, Calafell F, Gaspar C, Wong VC, Silveira I, Nicholson GA, et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. JAMA Neurol. 2007;64:1502–8.
Martins S, Soong BW, Wong VC, Giunti P, Stevanin G, Ranum LP, et al. Mutational origin of Machado–Joseph disease in the Australian Aboriginal communities of Groote Eylandt and Yirrkala. JAMA Neurol. 2012;69:746–51.
Costa IPD, Almeida BC, Sequeiros J, Amorim A, Martins S. A pipeline to assess disease-associated haplotypes in repeat expansion disorders: the example of MJD/SCA3 locus. Front Genet. 2019;10:38.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89.
Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Takezaki N, Nei M, Tamura K. POPTREE2: software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol Biol Evol. 2010;27:747–52.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Gaspar C, Lopes-Cendes I, Hayes S, Goto J, Arvidsson K, Dias A, et al. Ancestral origins of the Machado–Joseph disease mutation: a worldwide haplotype study. Am J Hum Genet. 2001;68:523–8.
Acknowledgements
The authors thank the Israeli Machado-Joseph Association and all patients and families for their participation in this study.
Funding
This work was financed by the FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020 Operacional Programme for Competitiveness and Internationalisation (POCI), Portugal 2020; by the project NORTE-01-0145-FEDER-000008, supported by the Norte Portugal Regional Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund; and by Portuguese funds through FCT—Fundação para a Ciência e a Tecnologia, Ministério da Ciência, Tecnologia e Inovação, in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01–0145-FEDER-007274). SM is funded by the FCT research contract IF/00930/2013.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharony, R., Martins, S., Costa, I.P.D. et al. Yemenite-Jewish families with Machado–Joseph disease (MJD/SCA3) share a recent common ancestor. Eur J Hum Genet 27, 1731–1737 (2019). https://doi.org/10.1038/s41431-019-0449-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0449-7
