
Abstract
High blood pressure (BP) is a major risk factor for cardiovascular disease (CVD) and is more prevalent in African Americans as compared to other US groups. Although large, population-based genome-wide association studies (GWAS) have identified over 300 common polymorphisms modulating inter-individual BP variation, largely in European ancestry subjects, most of them do not localize to regions previously identified through family-based linkage studies. This discrepancy has remained unexplained despite the statistical power differences between current GWAS and prior linkage studies. To address this issue, we performed genome-wide linkage analysis of BP traits in African-American families from the Family Blood Pressure Program (FBPP) and genotyped on the Illumina Human Exome BeadChip v1.1. We identified a genomic region on chromosome 1q31 with LOD score 3.8 for pulse pressure (PP), a region we previously implicated in DBP studies of European ancestry families. Although no reported GWAS variants map to this region, combined linkage and association analysis of PP identified 81 rare and low frequency exonic variants accounting for the linkage evidence. Replication analysis in eight independent African ancestry cohorts (N = 16,968) supports this specific association with PP (P = 0.0509). Additional association and network analyses identified multiple potential candidate genes in this region expressed in multiple tissues and with a strong biological support for a role in BP. In conclusion, multiple genes and rare variants on 1q31 contribute to PP variation. Beyond producing new insights into PP, we demonstrate how family-based linkage and association studies can implicate specific rare and low frequency variants for complex traits.
Similar content being viewed by others

Introduction
Elevated blood pressure (BP), or hypertension (HTN), through systolic (SBP) and/or diastolic (DBP) BP, is the most common cardiovascular condition across the global population, and the major risk factor for stroke, cardiovascular disease (CVD), renal failure, and congestive heart failure [1, 2]. Susceptibility to HTN varies considerably by age, sex, body mass index (BMI), ancestry, sodium intake, endothelial and vascular changes, stress, alcohol consumption, to name a few. However, the biological routes to subsequent disease through these risk factors remain obscure [3]. Moreover, there are significant population differences in HTN prevalence and risk, as exemplified by African Americans displaying the highest prevalence, and consequently, experiencing greater risk of CVD and end-stage renal disease as compared to their European Americans counterparts [1, 4]. BP is also a classic quantitative trait with an estimated heritability from family data that ranges from 30 to 40% [5]. There is currently optimism that identification of BP genes and their pathways will illuminate HTN and target organ damage causes, irrespective of whether these pathways are perturbed by genetic, lifestyle and/or environmental factors [6, 7]. Moreover, these BP pathways are expected to enable the targeted development of therapeutics.
Recent well-powered multi-ancestry GWAS or gene-centric studies have shed considerable insight into the underlying genetic basis of BP by identify several susceptibility loci [8,9,10,11]. However, the variance explained by the detected loci is less than 3.5% [8, 9, 11] of inter-individual variation in BP, supporting the hypothesis that rare and low frequency variants may contribute to BP variation. Multiple statistical methods for testing rare variant association have been developed for population-based samples [12,13,14,15,16] and family data [17,18,19,20]. It has long been known that family-based designs have an advantage in detecting rare variants [17, 19]. In the past two decades, before GWAS became popular, numerous large-scale genome-wide linkage studies on BP and HTN have been reported, including the well-designed studies by the NHLBI-supported Family Blood Pressure Program (FBPP) and the UK Medical Research Council British Genetics of Hypertension (BRIGHT) study [5, 21,22,23]. However, the linkage findings were few, often not having appropriate replication samples, and the observed linked loci did not, for the most part, overlap the subsequent GWAS-derived association loci. This has led to the view that while common variants are responsible for GWAS findings, the findings of linkage mostly arose from rare and low frequency variants. We sought to test this hypothesis directly using data on SBP and DBP variation.
Materials and methods
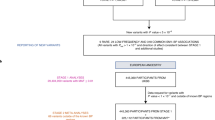
Our analysis flow chart is presented in Fig. 1. We first used primary exome array data on 1802 African American families from the Family Blood Pressure Program (FBPP) for linkage analysis (described in Supplementary Methods). The BP outcomes include the residues of SBP, DBP, and pulse pressure (PP), imputed for anti-hypertention medication, and adjusted for age and BMI. Population structure and family structure were modeled by the first ten principal components (PC) of genome and the kinship matrix (Supplementary Methods). Linkage analysis was performed on 20,791 Single Nucleotide Polymorphisms (SNPs) (minor allele frequency > 0.2 and linkage disequilibrium r2 < 0.1) using Merlin [24]. Significant linkage evidence was defined as LOD > 3 corresponding to a principled posterior probability of linkage of 95 percent [25, 26].

Analysis flow chart
In order to testify that our observed linkage evidence was driven by multiple low frequency/rare variants but not common variats, we performed a serious of diagnoses and simulations including: (1) estimated the heritability attributable to common variants using 6194 unrelated African Americans from the Candidate Gene Association Resource (CARe) consortium (Supplementary Methods); (2) estimated the regional heritability attributable to common variants in every 40 cM under the linkage peak and calculated its correlation with LOD score in the same region; and (3) simulated a quantitative trait influenced by single common variant (minor allele frequency [MAF] = 0.1 or 0.4 and accounting for 1% phenotypic variation) under the FBPP pedigree structure and perform linkage analysis using Merlin [24] for 100 times (Supplementary Methods).
We next examined all SNPs in the candidate linkage interval specified with boundary LOD > 1 and harboring the linkage peak. Risk variants were selected in three steps. In step 1, we performed family-based association analysis using a linear mixed model using ASSOC in S.A.G.E [27]. SNPs with association evidence were selected with either P ≤ 0.1 (marginal significance) or absolute regression coefficient beta ≥ 5 (large allelic effect). In step 2, we filtered the SNPs from step 1 for linkage contribution by presenting at least twice in a family with family-specific LOD ≥ 0.1. In step 3, we excluded intergenic and intronic SNPs, and SNPs in complete LD. We then defined a risk score for those risk SNPs as \(r_i = x_i^T\beta\), where β is the estimated regression coefficients of SNPs, and \(x_i^T\)is a (transposed) vector of the number of risk alleles carried by individual i. Linkage analysis was further performed conditional on this risk score.
Finally, we performed a family-based SKAT-O[28] test to investigate the cumulative effect of risk variants in FBPP. Variants were pooled together and grouped into non-overlapping windows in SKAT-O. Genetic pathway enrichment analysis of genes harboring risk variants was performed using the software package STRING [29], which implements a prediction pipeline for inferring protein-protein interactions (PPI). We reported gene sets that were either significantly co-expressed or involved in experimentally validated PPI with a medium score or higher (score ≥ 0.4). The enriched pathways and their involved genes were followed-up to group variants for SKAT-O test. SNP-set replication analysis was performed using 16,968 individuals with African ancestries collected from eight independent cohorts: the African American Diabetes Mellitus Study (AADM), the Atherosclerotic Risk in Communities Study (ARIC), the Mt. Sinai BioMe Biobank (BioMe), the Vanderbilt University Medical Center Biobank (BioVU), the Health and Retirement Study (HRS), the Howard University Family Study (HUFS), the Multi-Ethnic Study of Atherosclerosis (MESA), and the southwest Nigeria cohort collected by Loyola University at Chicago (Nigeria) (Supplementary Methods). Replication analysis was conducted in individual cohorts and meta-analyzed using Fisher’s method.
Results
Supplementary Table 1 presents the characteristics of the FBPP samples included. The residuals of SBP, DBP, and PP are all approximately normally distributed and required no further transformations (Supplementary Fig. 1). Linkage analysis demonstrated three significant linkage peaks with LOD > 3 on chromosomes 1, 17, and 19, all observed with PP. The largest linkage peak was at chromosome 1q31 (LOD = 3.8). Additional linkage evidence (LOD > 2) was observed on chromosomes 5, 19, and 20 for all three BP traits (Table 1, Supplementary Fig. 2).
We followed-up on the largest linkage peak at 1q31, which reached genome-wide statistical significance and overlaps with a linkage region already implicated in prior BP studies in the FBPP European ancestry families, and in other studies [30,31,32,33]. However, existing BP GWAS loci do not overlap with this region. We estimated the heritability attributable to common variants in every 40 cM across the genome in unrelated samples from CARe. The correlations between LOD scores and estimated regional heritabilities were consistently low and negative for all three BP traits (ρ = −0.021,−0.098 and −0.211 in SBP, DBP and PP; Supplementary Fig. 3). In particular, the estimated heritability at 1q31 was not statistically significant (h2 = 0.01% ± 0.44%) for PP. Our analysis consistently suggests that common variants do not account for observed linkage evidence for BP, including at 1q31 (Supplementary Table 2). Additionally, the heritability estimates are essentially unchanged for all BP traits on chromosome 1 with or without including the SNPs in the linkage region. Futhremore, we used simulations of a single common variant (Materials and Methods) to demonstrate that the observed linkage evidence could not be due to common variants. Among the 100 simulations, the maximum LOD scores were 2.31 and 2.28 for MAF 0.1 and 0.4, respectively, suggesting the common variants cannot explain the linkage evidence observed on 1q31.
Informed by the linkage evidence, we focus on PP trait in the following analysis. To search for specific variants that can explain the linkage evidence, we considered the region with boundaries LOD > 1 harboring the linkage peak (171.1–203.9 Mb; Fig. 2), a region that contained 2533 variants on the exome array. A total of 81 exonic SNPs were left after filtering by association evidence and linkage contribution (Materials and Methods; Supplementary Table 3). Linkage analysis conditional on the risk score defined by these 81 SNPs did not change the linkage peak location but reduced the LOD score from 3.8 to 1.2 (Fig. 2), strongly suggesting that these variants do explain a majority of the original PP linkage evidence.

Significant evidence for linkage of PP to human chromosome 1 from the FBPP study. The linkage evidence is plotted conditional on, or not, with respect to the risk score calculated by the 81 variants within the region. The 81 variants within 54 genes are also shown
We next performed family-based SKAT-O [28] tests for the pooled 81 exonic variants in FBPP samples. The observed P value is small for PP, as well as for SBP and DBP, which is not surprising because of selected SNPs (Table 2). We then performed SKAT-O analysis for PP with the same 81 SNPs in eight replication samples separately and the results then meta-analyzed (Table 2). We observed a marginally significant association for PP (P = 0.0509) and the corresponding association evidence of SBP, DBP was marginal (P = 6.41 × 10−2 and 9.11 × 10−2, respectively).
The 81 variants within the linkage interval are spread out across numerous genes and, so for genetic specificity, we divided the variants into 20 non-overlapping windows of four variants each (the last window has five variants) (Supplementary Table 3). SKAT-O tests were performed for each window (Supplementary Table 4), and the most significant window for PP consists of four variants (rs148482637:C>T, rs35946265:T>C, rs77277070:T>C, and rs112997656:G>A) on three genes: ASTN1 (MIM:600904), SEC16B (MIM:612855) and C1orf49 (MIM:611154), with P value 2.43 × 10−4 in FBPP, and this window was significant in the meta-analysis of the replication cohorts (P = 4.16 × 10−2, Table 3). The corresponding results for SBP and DBP were also presented in Table 3.
A second approach to assess the veracity of the linkage evidence is to assess the functions encoded by the 54 genes with the 81 variants. The overall protein-protein interaction (PPI) enrichment P value for the 54 genes is 1.27 × 10−7. We identified one GO biological process, one GO molecular function and one Kegg pathway that were significantly enriched (Table 4). The GO biological pathways “termination of G-protein coupled receptor signaling” (GO:0038032) involves four regulators of G-Protein Signaling: RGSL1 (MIM:611012), RGS21 (MIM:612407), RGS1 (MIM:600323), RGS13 (MIM:607190). The four variants in these four genes (rs7535533:A>G, rs77664911:C>T, rs144803737:T>A, and rs143481310:C>T) are associated with with PP in FBPP (SKAT-O P = 0.018) but could not be replicated (Table 4, P = 0.659). The GO molecular functions “N,N-dimethylaniline monooxygenase activity” (GO:0004499) involves flavin-containing monooxygenases: FMO1 (MIM:136130), FMO2 (MIM:603955), and FMO3 (MIM:136132). The four variants in these three genes (rs2266782:G>A, rs200985584:G>A, rs2020866:C>T, and rs149030329:G>A) are associated with PP in FBPP (SKAT-O P = 0.026) and the association is significant in the replication cohorts (P = 0.027). The extracellular matrix (ECM) receptor interaction (hsa4512) enriched in KEGG includes four genes: TNN, TNR (MIM:601995), LAMC1 (MIM:150290), LAMC2 (MIM:150292). The six variants in these four genes (rs143192203:G>T, rs859427:T>C, rs148657090:A>T, rs141408335:C>T, rs143817389:A>G, and rs35281117:T>A) are associated with PP in FBPP (SKAT-O P = 0.022) but fails to replicate (P = 0.459). Again we also reported the corresponding association evidence for SBP and DBP in Table 4.
Gene expression analyses of multiple tissues [34] on genes of interests in the most significant window and significant pathways are shown in Supplementary Figs. 4–7 by looking at the GTEx database.
Discussion
As GWAS have become a popular approach to detect genetic variants underlying complex traits, linkage analysis has become less popular, even ignored, since it is considered to be of low statistical power. However, the genetic variants identified by BP GWAS do not always localize to the genomic regions identified in previous linkage studies [21,22,23, 35,36,37,38], suggesting that the latter might provide novel clues to BP genetic susceptibility. A possible explanation for these discrepant findings is that GWAS are targeting common variants but have little power when multiple rare variants in a region (gene) contribute to trait variation, as can be detected with family-based linkage studies [17]. Thus, a search for multiple rare or low frequency BP variants in linkage regions may be fruitful. The success rate of this general strategy is, however, unknown.
In this study, we assessed such a BP linkage region using African American families from the FBPP study and who have been genotyped using the exome array, a platform specifically designed for examining functional variants with variants enriched for many cardiovascular traits including BP. We identified seven genomic regions linked to BP traits (Table 1) with LOD score > 2 and three with significant linkage signals (LOD > 3). Interestingly, the most significant evidence came from a locus for PP on human chromosome 1q31 (LOD score = 3.8), previously observed for SBP and DBP in European Americans [30, 32, 33, 39] but not enriched for BP GWAS signals [35,36,37,38, 40, 41]. Since this locus has shown significant linkage evidence in multiple studies of multiple ancestries it remains as a credible BP locus whose genetics need to be dissected. The heritability analysis in 6194 unrelated African American subjects also concluded that common variants in this region are unlikely to account for BP variation (Supplementary Table 2). Our computer simulations corroborated that even a common variant accounting for 1% phenotypic variation is unable to produce the observed linkage evidence (Materials and Methods), suggesting that multiple rare and low frequency variants are likely to be the causes of the linkage evidence. Our association analysis was focus on PP because of the observed linkage evidence in the chromosome 1 region and identified 81 exonic variants (Supplementary Table 3) that explained most of the observed PP linkage evidence (Fig. 1). Not surprisingly, most of these 81 variants are low frequency or rare functional variants. Nevertheless, we were able to replicate the association evidence for these 81 variants in the meta-analysis of eight additional African ancestry cohorts with marginally significance (P = 0.0509, Table 2). Because we tested the 81 variants together in replication cohorts, there was no need to adjust for multiple comparisons. Using the genetic score defined by the 81 variants, we observed a substantial drop of LOD score (from 3.8 to 1.2), indicating that the selected variants explained substantial linkage evidence on chromosome 1. This study included the largest exome-array replication analysis in African Americans. Although the replication sample size (N = 16,968) was still much smaller than the large exome-array study of adult height (N = 59,804) [42], we observed the same degree of replication evidence (P values were between 0.05 and 0.01 without adjusting for multiple tests). Our and other’s studies clearly suggest that the replication analyses for rare variant associations are facing a big challenge.
The main challenge is to identify which of the 54 genes, and their corresponding variants, within the linkage interval are responsible for inter-individual PP variation. Because the 81 variants are sparsely located in 54 genes, we performed non-overlapping window SKAT-O analysis to reduce the number of comparisons although a sliding window analysis had essentially the same result. We demonstrated that one interval of four variants with the genes ASTN1, SEC16B, and C1orf49 is the most significantly associated in our discovery with PP and is also replicated (Table 3). However further data on new samples are needed to be assured of their significance. These three genes harbor four non-synonymous variants and demonstrate a consistent protective effect on PP in the FBPP. ASTN1 and its two upstream genes RFWD2 and PAPPA2 have been shown to be strongly expressed in the renal outer medulla and show protective effects on HTN in Dahl S rats [43, 44], consistent with what we observed with humans in this study. This effect may be mediated through pathways that affect BP via renal transcellular Na and K electrochemical gradients and tubular Na transport, mitochondrial TCA cycle and cell energetics, and circadian rhythms [43]. For humans, we used the GTEx database [34] to search for the expression domains of the four genes: ASTN1 is mainly expressed in the brain; and, SEC16B is highly expressed in the kidney and liver (Supplementary Fig. 4). Therefore, these four genes are highly relevant candidate BP genes. However, functional tests are needed to confirm these findings.
To assess function, we also examined protein-protein interaction data for the 54 genes using STRING [29] and observed significant enrichment in two GO and one Kegg pathways (Table 4). The GO molecular function pathway “N,N-dimethylaniline monooxygenase activity” (GO:004499) involves flavin-containing monooxygenases (FMOs) genes: FMO1, FMO2 and FMO3 (Table 4). The four exonic variants in these genes are associated with PP in both FBPP and replication cohorts (Table 4). FMO3 is involved in the formation of trimethylamine (TMA) N-oxides (TMAO) [45]. Organic osmolytes (i.e., TMAO) can counteract an increase in osmotic pressure and peripheral resistance, and hypertension can develop when there are no organic osmolytes [46]. Individuals with deficient FMO3 activity have a higher prevalence of HTN and other CVD, because of a decrease in formation of TMAO to counterbalance the effects of a higher osmotic pressure and peripheral resistance [47]. FMO1 is highly expressed in kidney, FMO2 is highly expressed in lung, aorta and adipose and FMO3 is highly expressed in liver and adipose (Supplementary Fig. 5). Therefore, the FMO family may regulate BP through these pathways but the specific functional deficits remain unknown.
These results, taken together, support the hypothesis that multiple rare and low frequency variants may underlie linkage peaks. If so, these regions represent new opportunities for gene discovery. This hypothesis immediately suggests many corollaries that can be tested: (1) rare functional variants can also have small-to-modest genetic effects and behave in a non-Mendelian manner, (2) the clustering of variants within the linkage region can occur from diverse genetic effects at a single gene (intragenic allelic heterogeneity) or from the effects of multiple variants in different genes (intergenic allelic heterogeneity), and (3) when different genes are implicated, as in here, they do not necessarily have to have the same function but need to perturb the trait through different pathways. These predictions depend on how gene function is distributed in the human genome. In contrast, it is surprising that the allelic effects are small to modest as for GWAS rather than being large as for coding variants in rare disease. This aspect may simply reflect the biology of a synthetic trait like BP that represents the effects of many genes and numerous independent pathways, but none with overwhelming importance. Perhaps, major physiological traits under homeostatic control have complex distributed regulation so that there is no single point of vulnerability. Consistent with this evidence, to date, variants in Mendelian HTN syndromes, do not explain the variance of BP in the general population.
Although linkage studies are often considered to have low statistical power, many disease variants have been identified through linkage analysis, such as the BRCA1 and BRCA2 genes for breast cancer and RBFOX1 for BP [48, 49]. We used LOD score > 3, the initial proposal by Morton [25], as our genome-wide significance level corresponding to a principled posterior probability of linkage of 95 percent (Ott 1991 [26], p.66). Our linkage peak on chromosome 1 with LOD = 3.8 is also well above the threshold of 3.3 suggested by Lander and Kruglyak [50]. The linkage evidence at this region has been replicated multiple times in independent studies [30, 32, 33, 39]. We further calculated statistical power. The power to achieve a LOD score of 3.0 is 49.5% for a multiple point linkage analysis assuming a model with 5% quantitative trait loci (QTL) additive variance, 60% residual shared variance, no recombination between marker and the QTL, and a sample size of 4394 with sibship size 3 (the average sibship size of FBPP). However, the FBPP study was designed to enrich for families with HTN genes by over-sampling high BP family members such that our power could be underestimated.
Linkage analysis is not affected by allelic heterogeneity but association analysis is. Thus a reasonable hypothesis is that the observed significant linkage evidence on chromosome 1 is driven by multiple low frequency or rare variants, which is the focus of our study. In addition, we calculated the BP variation explained by common variants across the genome using the summary statistics from the large BP GWAS study in ICBP [36]. The estimated variation explained by common variants is between 11.62 and 12.13%, which is much lower than the heritability estimated from family data (~35% [5]). Thus, our hypothesis that the linkage peak is driven by multiple low frequency or rare variants is reasonable.
In conclusion, our linkage analysis identified a genome wide significant region on 1q31 harboring PP variants. Follow-up association revealed multiple exonic variants in this region contributing the observed linkage evidence and the association evidence was replicable in independent cohorts. Association analysis and pathway analysis further identified multiple genes, ASTN1, SEC16B, C1orf49, FMO1, FMO2, and FMO3 are associated with PP. Beyond producing new insights into BP, we demonstrate how family-based linkage and association studies can implicate specific rare and low frequency variants for complex traits.
References
Berry JD, Dyer A, Cai X, Garside DB, Ning H, Thomas A, et al. Lifetime risks of cardiovascular disease. New Engl J Med. 2012;366:321–9.
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60.
Ehret GB, Caulfield MJ. Genes for blood pressure: an opportunity to understand hypertension. Eur Heart J. 2013;34:951–61.
Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr., et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560–72.
Cooper RS, Luke A, Zhu X, Kan D, Adeyemo A, Rorimi C, et al. Genome scan among Nigerians linking blood pressure to chromosomes 2, 3, and 19. Hypertension. 2002;40:629–33.
Ehret GB. Genome-wide association studies: contribution of genomics to understanding blood pressure and essential hypertension. Curr Hypertens Rep. 2010;12:17–25.
Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56.
Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet. 2016;48:1171–84.
Surendran P, Drenos F, Young R, Warren H, Cook JP, Manning AK, et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat Genet. 2016;48:1151–1161.
Liu C, Kraja AT, Smith JA, Brody JA, Franceschini N, Bis JC, et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat Genet. 2016;48:1162–70.
Hoffmann TJ, Ehret GB, Nandakumar P, Ranatunga D, Schaefer C, Kwok PY, et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat Genet. 2017;49:54–64.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–75.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384.
Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21.
Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86:832–8.
Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–67.
Zhu X, Feng T, Li Y, Lu Q, Elston RC. Detecting rare variants for complex traits using family and unrelated data. Genet Epidemiol. 2010;34:171–87.
Feng T, Elston RC, Zhu X. Detecting rare and common variants for complex traits: sibpair and odds ratio weighted sum statistics (SPWSS, ORWSS). Genet Epidemiol. 2011;35:398–409.
Epstein MP, Duncan R, Ware EB, Jhun MA, Bielak LF, Zhao W, et al. A statistical approach for rare-variant association testing in affected sibships. Am J Hum Genet. 2015;96:543–54.
Wang H, Cade BE, Chen H, Gleason KJ, Saxena R, Feng T, et al. Variants in angiopoietin-2 (ANGPT2) contribute to variation in nocturnal oxyhaemoglobin saturation level. Hum Mol Genet. 2016;25:5244–53.
Province MA, Kardia SL, Ranade K, Rao DC, Thiel BA, Cooper RS, et al. A meta-analysis of genome-wide linkage scans for hypertension: the National Heart, Lung and Blood Institute Family Blood Pressure Program. Am J Hypertens. 2003;16:144–7.
Caulfield M, Munroe P, Pembroke J, Samani N, Dominiczak A, Brown M, et al. Genome-wide mapping of human loci for essential hypertension. Lancet. 2003;361:2118–23.
Wu X, Kan D, Province M, Quertermous T, Rao DC, Chang C, et al. An updated meta-analysis of genome scans for hypertension and blood pressure in the NHLBI Family Blood Pressure Program (FBPP). Am J Hypertens. 2006;19:122–7.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101.
Morton NE. Sequential tests for the detection of linkage. Am J Human Genet. 1955;7:277–318.
Ott J. Analysis of human genetic linkage. Rev. edn. Baltimore: Johns Hopkins University Press; 1991.
Elston RC, Gray-McGuire C. A review of the ‘Statistical Analysis for. Genet Epidemiology’ (S A G E) Softw Package Human Genom. 2004;1:456–9.
Chen H, Meigs JB, Dupuis J. Sequence kernel association test for quantitative traits in family samples. Genet Epidemiol. 2013;37:196–204.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRINGv10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52.
Chang YP, Liu X, Kim JD, Ikeda MA, Layton MR, Weder AB, et al. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet. 2007;80:253–64.
Perola M, Sammalisto S, Hiekkalinna T, Martin NG, Visscher PM, Montgomery GW, et al. Combined genome scans for body stature in 6,602 European twins: evidence for common Caucasian loci. PLoS Genet. 2007;3:e97.
Hunt SC, Ellison RC, Atwood LD, Pankow JS, Province MA, Leppert MF. Genome scans for blood pressure and hypertension: the National Heart, Lung, and Blood Institute Family Heart Study. Hypertension. 2002;40:1–6.
James K, Weitzel LR, Engelman CD, Zerbe G, Norris JM. Framingham Heart S: Genome scan linkage results for longitudinal systolic blood pressure phenotypes in subjects from the Framingham Heart Study. BMC Genet. 2003;4(Suppl 1):S83.
Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–87.
Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9.
Franceschini N, Fox E, Zhang Z, Edwards TL, Nalls MA, Sung YJ, et al. Genome-wide association analysis of blood-pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am J Hum Genet. 2013;93:545–54.
Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Human Genet. 2015;96:21–36.
Perola M, Kainulainen K, Pajukanta P, Terwilliger JD, Hiekkalinna T, Ellonen P, et al. Genome-wide scan of predisposing loci for increased diastolic blood pressure in Finnish siblings. J Hypertens. 2000;18:1579–85.
Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–76.
Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43:531–8.
Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. 2017;542:186–90.
Cowley AW Jr., Moreno C, Jacob HJ, Peterson CB, Stingo FC, Ahn KW, et al. Characterization of biological pathways associated with a 1.37 Mbp genomic region protective of hypertension in Dahl S rats. Physiol Genom. 2014;46:398–410.
Cowley AW Jr, Yang C, Kumar V, Lazar J, Jacob H, Geurts AM, et al. Pappa2 is linked to salt-sensitive hypertension in Dahl S rats. Physiol Genom. 2016;48:62–72.
Cashman JR, Camp K, Fakharzadeh SS, Fennessey PV, Hines RN, Mamer OA, et al. Biochemical and clinical aspects of the human flavin-containing monooxygenase form 3 (FMO3) related to trimethylaminuria. Curr Drug Metab. 2003;4:151–70.
Lifton RP. Molecular genetics of human blood pressure variation. Science. 1996;272:676–80.
Treacy EP, Akerman BR, Chow LM, Youil R, Lin CB, Bruce AG, et al. Mutations of the flavin-containing monooxygenase gene (FMO3) cause trimethylaminuria, a defect in detoxication. Hum Mol Genet. 1998;7:839–45.
Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Human Genet. 1998;62:676–89.
He KY, Wang H, Cade BE, Nandakumar P, Giri A, Ware EB, et al. Rare variants in fox-1 homolog A (RBFOX1) are associated with lower blood pressure. PLoS Genet. 2017;13:e1006678.
Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–7.
Funding
This work was supported in part by NIH grants HL086694 to AC and HG003054 to XZ. TLE was supported by NIH/NHLBI grant HL121429, JNH was supported by T32CA160056, RJFL was support by NIH grants DK107786, DK110113, and U01HG007417. The GenNet study is funded by NIH grants HL086694. The Genetic Epidemiology Network of Arteriopathy (GENOA) was supported by the National Heart, Lung and Blood Institute (HL054464, HL054457, HL054481, HL087660, HL086694, HL119443) of the National Institutes of Health. Genotyping was performed at the Center for Inherited Disease Research (CIDR). The HyperGEN cohort study is funded by cooperative agreements (U10) with NHLBI: HL54471, HL54472, HL54473, HL54495, HL54496, HL54497, HL54509, HL54515, and 2 R01 HL55673-12. The study involves: University of Utah (Network Coordinating Center, Field Center, and Molecular Genetics Lab); University of Alabama at Birmingham (Field Center and Echo Coordinating and Analysis Center); Medical College of Wisconsin (Echo Genotyping Lab); Boston University (Field Center); University of Minnesota (Field Center and Biochemistry Lab); University of North Carolina (Field Center); Washington University (Data Coordinating Center); Weil Cornell Medical College (Echo Reading Center); National Heart, Lung, & Blood Institute. For a complete list of HyperGEN Investigators: http://www.biostat.wustl.edu/hypergen/Acknowledge.html. The Supports for replication cohorts are shown in Supplementary.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wang, H., Nandakumar, P., Tekola-Ayele, F. et al. Combined linkage and association analysis identifies rare and low frequency variants for blood pressure at 1q31. Eur J Hum Genet 27, 269–277 (2019). https://doi.org/10.1038/s41431-018-0277-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0277-1
This article is cited by
-
Rare coding variants in RCN3 are associated with blood pressure
BMC Genomics (2022)
-
Identifying Rare Variant Associations in Admixed Populations
Scientific Reports (2019)
