
Abstract
In most patients with intellectual disability (ID), the etiology is unknown, but lately several de novo variants have been associated with ID. One of the involved genes, CUX2, has twice been reported to be affected by a de novo variant c.1768G>A; p.(Glu590Lys) in patients with ID or epileptic encephalopathy. CUX2 is expressed primarily in nervous tissues where it may act as a transcription factor involved in neural specification. Here we describe a third case who was diagnosed with epilepsy including general and myoclonic seizures, moderate to severe cognitive disability, and infantile autism. The patient was heterozygous for the c.1768G>A; p.(Glu590Lys) variant in CUX2 identified by whole exome sequencing. These findings strongly suggest a causal impact of this variant and add to our understanding of a subset of patients with ID, seizures, and autism spectrum disorder as well as suggest an important role for the CUX2 gene in human brain function.
Similar content being viewed by others

Introduction
Intellectual disability (ID) and autism spectrum disorder (ASD) are common clinical manifestations of congenital neurodevelopmental disorders with an estimated prevalence of 1.83% and 1.46%, respectively [1, 2]. In addition to the obvious handicap of being intellectually disabled, 22% of patients with ID also suffer from epilepsy, often beginning in the childhood [3]. The conditions are ascribed to various genetic abnormalities, which in multiple cases comprise de novo variants [4,5,6]. In around 30% of patients with genetically heterogenous disorders such as ID, the disorder can be genetically diagnosed by means of diagnostic exome sequencing when traditional technologies are uninformative [7]. In one case, a de novo variant was identified in CUX2 (c.1768G>A; p.(Glu590Lys)) (synonyms: CUTL2, KIAA0293) in a cohort study of 264 patients with epileptic encephalopathy (EE), and a further patient with the same variant has been described in another cohort study of 51 patients with ID [6, 8]. Neither of the studies presented detailed clinical data. Here we report a third case with the variant with ID, seizures, and ASD, which are the common phenotypic traits in the three cases.
Case presentation and methods
The proband is a female, the second child of healthy non-consanguineous parents, the mother was 32 years old and the father, 39 years old; the mother reported a medically uneventful pregnancy. The proband was born at term through uncomplicated vaginal delivery. There were no neonatal complications and no craniofacial dysmorphology was noted. At 6 months of age, the mother observed delayed development of the proband, especially lack of eye contact and head control. At the age of 4 years, a neuropsychological assessment was done, and the proband was diagnosed with moderate to severe ID and infantile autism. Motor development was also delayed, and she was described as having a rigid movement pattern with stereotypic movements, walking on toes and difficulties in getting up from a lying position. She had the onset of epilepsy at the age of 12 months with generalized and myoclonic seizures, which appeared to be provoked by stress and lack of sleep. The generalized seizures were described as episodes lasting 2–3 min, both during day and night, with unconsciousness, jerks, and twitching of the upper limbs. The myoclonic seizures were characterized by intensifying axial spasms up to several times daily. The proband was treated with lamotrigine and valproate resulting in the reduction of seizure frequency, and the treatment was discontinued at the age 13 of years with only occasional relapses. Additional findings included a tendency to hypersalivation and constipation. By late adolescence, she had no language and was deeply dependent on her parents. The proband’s family history included two intellectually disabled family members: her paternal grandmother’s cousin’s son, who was intellectually disabled and dysmorphic attributed to a chromosome 1/18 translocation and her maternal grandfather’s cousin’s daughter’s child who was diagnosed with ID and infantile autism of unknown etiology.
Urinalysis and analysis of lysosomal enzymes, long chain fatty acids, and phytanic acid as well as array-based comparative genomic hybridization and sequencing of MECP2 and CDKL5 showed normal results.
Exome sequencing was performed by the Ion AmpliSeq™ exome RDY kit (Life Technologies) for amplification of target regions (>97% of the coding region). Ion Chef was used to prepare libraries, which were sequenced using the Ion Proton system (Life Technologies). Base calling, pre-processing of the reads, short read alignment, and variant calling were performed using the Torrent Suite including the Torrent Variant Caller (TVC, Version 4.4–5.0) (Life Technology). Variant prioritization was performed with a cascade of filtering steps. Alamut Visual v.2.8 was used for the annotation and evaluation of missense, nonsense, splice site, and small indels variants. Finally, selected variants were validated by PCR and Sanger sequencing according to standard protocols to exclude technical artifacts and to test for segregation.
The clinical and variant details have been submitted to LOVD (https://databases.lovd.nl/shared/individuals/00154932).
Results
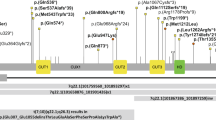
Whole exome sequencing revealed that the proband was heterozygous for c.1768G>A; p.(Glu590Lys) in the CUX2 gene (NM_015267.3), which was not identified in DNA from blood samples from her parents. The variant is not found among 245,000 alleles in the GnomAD database (http://gnomad.broadinstitute.org/), and it is predicted damaging in silico by SIFT (SIFT score 0) (http://sift.jcvi.org/www/SIFT), probably damaging in Polyphen2 (PPH2 score 0.999) (http://genetics.bwh.harvard.edu/pph2/), and disease-causing by MutationTaster (http://www.mutationtaster.org/). The specific variant has previously been reported in two patients with EE or ID [6, 8]. Their clinical data are summarized in Table 1.
Discussion
Several parameters make a causal relationship between the CUX2 variant and the proband’s phenotype with EE and ID highly probable: (1) the variant has been found de novo in three independent cases with EE or ID including the patient reported here; (2) the variant was absent from 245,000 alleles in the gnomAD database; (3) the variant is predicted in silico to affect CUX2 function; and (4) CUX2 likely has an important function in neural development and homeostasis.
Only sparse clinical information was available for the two reported patients with the CUX2 variant (Table 1) [6, 8]. For patient 1, the only information was that the patient had EE, and ID or ASD. The authors did not state that the variant was the cause of EE in the patient, but none of the other de novo variants identified in the patient were predicted to affect function. Patient 2 had a similar phenotype to our patient, with moderate to severe ID, motor retardation, and autistic features. His epilepsy consisted of benign neonatal seizures with onset at the age of 6 months, whereas our patient had myoclonic and generalized seizures from the age of 12 months. The patient reported here was described with a “Rett-like” phenotype in the patient files, and sequence analysis of MECP2 and CDKL5 was performed. However, she fulfilled only one of the criteria for Rett syndrome (stereotypic movements), so at the moment, there is no evidence that CUX2-related ID is associated with a Rett-like phenotype [9]. An additional patient with a de novo nonsense CUX2 variant (c.1897C>T, p.(Arg633*)) has been reported in a cohort study of patients with ASD, but no other clinical data were available [10].
The identification of three patients with a recurrent de novo variant makes it highly likely that the affected nucleotide, c.1768G, is a mutational hot-spot.
The CUX2 gene was first described in 1996 by Quaggin et al. [11] and is a member of a family of genes coding for transcription factors in metazoans involved in the control of proliferation and differentiation (reviewed in [12]). In higher vertebrates, there are two CUX genes, CUX1 and CUX2 [13], characterized by the presence of three highly similar regions called Cut 1, 2, and 3. CUX1 has not been linked to disease in humans. CUX2 is located on chromosome 12q23.13 and encodes a protein of 1486 amino acids. CUX2 contains several DNA binding domains and is likely to act as a transcription factor involved in the neural specification (http://www.uniprot.org/uniprot/O14529). In adult mice, CUX2 is predominantly expressed in neurons in the thalamus and limbic system, and in mouse embryos, CUX2 is expressed in the developing central and peripheral nervous systems supporting an important role of CUX2 in brain function [11]. Moreover, CUX1 and CUX2 are additive and complement each other to establish the final pattern of the dendritic tree and the number and strength of the synapses in layer II–III neurons of the cerebral cortex [14, 15]. More specifically, modulation of the function of CUX1 results in a notable effect on the development of the basal compartment of the dendrites whereas changes in CUX2 have a stronger influence on the apical compartment. These distinctive features of the CUX genes suggest that they might play a role in the functional diversification of layer II–III neurons into different subpopulation [16]. Furthermore, working memory is severely impaired in Cux2−/− mice which indicates that Cux2 influences circuits involved in cognition with potential implications for Cux2 in human disorders [15]. The CUX2 variant reported here leads to change of a glutamine, which is located in the Cut 1 repeat of CUX2. Ectopic expression of various recombinant proteins containing CUX2 Cut repeats was shown to accelerate DNA repair, and in vitro, Cut repeats from CUX2 stimulated the activity of 8-oxoguanine DNA glycosylase 1, which is involved in base excision repair [17]. CUX2 knockdown was shown to result in increased oxidative DNA damage in cortical neurons and in HCC38 breast tumor cells. Taken together, these results suggest that CUX2, like CUX1, functions as an accessory factor in base excision repair of oxidative DNA damage [17]. It is not known, if the variant reported here is associated with loss-of-function or gain-of-function, but it could be speculated that a variant affecting the function of a Cut repeat leads to oxidative DNA damage.
In conclusion, we here report the third patient with the de novo variant c.1768G>A; p.(Glu590Lys) in CUX2, strongly suggesting the role of CUX2 as a gene associated with ID, seizures, and ASD. Future studies will establish the frequency of ID caused by this particular variant and whether other CUX2 variants can cause a similar phenotype.
References
Christensen DL, Baio J, Van Naarden Braun K, Bilder D, Charles J, Constantino JN, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years--autism and developmental disabilities monitoring network, 11 sites, United States, 2012. Morb Mortal Wkly Report Surveill Summ. 2016;65:1–23.
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32:419–36.
Robertson J, Hatton C, Emerson E, Baines S. Prevalence of epilepsy among people with intellectual disabilities: a systematic review. Seizure. 2015;29:46–62.
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med: Off J Am Coll Med Genet. 2015;17:578–86.
Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21.
Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–50.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15.
Quaggin SE, Heuvel GB, Golden K, Bodmer R, Igarashi P. Primary structure, neural-specific expression, and chromosomal localization of Cux-2, a second murine homeobox gene related to Drosophila cut. J Biol Chem. 1996;271:22624–34.
Nepveu A. Role of the multifunctional CDP/Cut/Cux homeodomain transcription factor in regulating differentiation, cell growth and development. Gene. 2001;270:1–15.
Neufeld EJ, Skalnik DG, Lievens PM, Orkin SH. Human CCAAT displacement protein is homologous to the Drosophila homeoprotein, cut. Nat Genet. 1992;1:50–5.
Cubelos B, Nieto M. Intrinsic programs regulating dendrites and synapses in the upper layer neurons of the cortex. Commun Integr Biol. 2010;3:483–6.
Cubelos B, Sebastian-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, et al. Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron. 2010;66:523–35.
Cubelos B, Briz CG, Esteban-Ortega GM, Nieto M. Cux1 and Cux2 selectively target basal and apical dendritic compartments of layer II-III cortical neurons. Dev Neurobiol. 2015;75:163–72.
Pal R, Ramdzan ZM, Kaur S, Duquette PM, Marcotte R, Leduy L, et al. CUX2 protein functions as an accessory factor in the repair of oxidative DNA damage. J Biol Chem. 2015;290:22520–31.
Acknowledgments
We thank the family for their participation in the study. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interests.
Informed consent
Informed consent was obtained from both parents as the patient was under 18 years of age.
Rights and permissions
About this article

Cite this article
Barington, M., Risom, L., Ek, J. et al. A recurrent de novo CUX2 missense variant associated with intellectual disability, seizures, and autism spectrum disorder. Eur J Hum Genet 26, 1388–1391 (2018). https://doi.org/10.1038/s41431-018-0184-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0184-5
This article is cited by
-
Single-nucleus gene and gene set expression-based similarity network fusion identifies autism molecular subtypes
BMC Bioinformatics (2023)
-
CUX2 deficiency causes facilitation of excitatory synaptic transmission onto hippocampus and increased seizure susceptibility to kainate
Scientific Reports (2022)
