
Abstract
Tubulinopathies are a heterogeneous group of conditions with a wide spectrum of clinical severity resulting from variants in genes of the tubulin superfamily. Variants in TUBG1 have been described in three patients with posterior predominant pachygyria and microcephaly. We here report eight additional patients with four novel heterozygous variants in TUBG1 identified by next-generation sequencing (NGS) analysis. All had severe motor and cognitive impairment and all except one developed seizures in early life. The core imaging features included a pachygyric cortex with posterior to anterior gradient, enlarged lateral ventricles most pronounced over the posterior horns, and variable degrees of reduced white matter volume. Basal ganglia, corpus callosum, brainstem, and cerebellum were often normal, in contrast to patients with variants in other tubulin genes where these structures are frequently malformed. The imaging phenotype associated with variants in TUBG1 is therefore more in line with the phenotype resulting from variants in LIS1 (a.k.a. PAFAH1B1). This difference may, at least in part, be explained by gamma-tubulin’s physiological function in microtubule nucleation, which differs from that of alpha and beta-tubulin.
Similar content being viewed by others
Introduction
The gamma-tubulin protein (TUBG1) was first described by Oakley and Oakley [1], and the encoding TUBG1 gene has later been mapped to chromosome 17q2 [2,3,4]. TUBG1 shares 94.6% of nucleotides and 97.3% of amino acids with its paralog TUBG2, but despite this high degree of similarity they appear not to be functionally synonymous. Both TUBG1 and TUBG2 are highly expressed in the developing and mature human brain, including the cerebral cortex, cerebellum, thalamus, and hippocampus, with TUBG1 being expressed more abundantly than TUBG2 [5].
Variants in genes belonging to the tubulin superfamily, including TUBA1A, TUBB2A, TUBB2B, TUBB3, TUBB, and TUBG1, have been associated with a spectrum of cortical malformations through disruption of normal microtubule interactions, which are involved in neuronal cell proliferation, migration and differentiation, as well as axon growth and guidance [6,7,8,9,10,11,12,13,14,15]. Microtubule nucleation precedes the formation of bipolar spindles and separation of chromosomes in mitosis, steps that are necessary for the progression of the cell cycle [16, 17]. Unlike alpha-tubulin and beta-tubulin, gamma-tubulin is not incorporated in the microtubule lattice but is required for the polymerization of the alpha-tubulin and beta-tubulin proteins. Therefore, gamma-tubulin localizes to the centrosome during interphase. This process is mediated by protein kinases [18]. Two gamma-tubulin proteins associate with gamma complex proteins 2 and 3 (GCP2, GCP3) to form a gamma-tubulin small complex (γTuSC). Binding of GCP4, GCP5, and GCP6 to several copies of γTuSC results in the formation of a γ-tubulin ring complex (γTuRC). γTuRC links microtubules to the spindle pole during mitosis [19]. Insufficient proliferation due to defective microtubule function can ultimately lead to microcephaly [8]. Defects in neuronal proliferation and migration are linked to the role microtubules play in cell shape and orientation [20].
Protein structures within the tubulin superfamily show a high degree of similarity. However, the phenotypic differences associated with variants in the various tubulin isotypes support the hypothesis that each tubulin has a distinctive function [21].
An alanine-scanning mutagenesis screen of human gamma-tubulin in S. pombe showed that all deleterious variants in the TUBG1 gene were found in residues predicted to be located at the surface, some in positions to interact with alpha and/or beta-tubulin at the microtubule lattice. The localization of these variants might therefore indicate domains within the protein that are responsible for gamma-tubulin’s individual function [22, 23].
To date, three unrelated patients with de novo variants in TUBG1 have been reported [14]. Two had microcephaly and bilateral symmetric pachygyria with a posterior to anterior gradient on imaging. They suffered from spastic quadriparesis and were bedridden. The third patient presented with a milder phenotype with normal head circumference, mild intellectual disability, and posterior pachygyria. The corpus callosum was malformed in all three patients, whereas the basal ganglia, the cerebellum, and the brainstem were spared. All three patients presented with seizures in early life.
We report the identification of eight additional patients from seven families with four novel heterozygous variants in TUBG1, contributing to a further delineation of the associated clinical and imaging phenotype.
Methods
Patient samples
Patients were recruited through the international research network of the authors. The study was approved by the Institutional Review Board of the UZ Brussel (B.U.N. 143201214360), the Institutional Review Board at Seattle Children’s Hospital, and the Pediatric Ethics Committee of the Tuscany Region. Informed consent was obtained from all families prior to genetic studies. Clinical data were collected through clinical examination and review of medical records. All imaging data were reviewed by AJ, NDD, and WBD.
Blood samples for DNA preparation and genetic investigation were obtained with informed consent from patients and parents. DNA was extracted using standard protocols.
For patients 1 and 2, variant analysis was performed using gene panel analysis. This analysis was performed at the Center of Medical Genetics, UZ Brussel in collaboration with the Brussels Interuniversity Genomics High Throughput core (BRIGHT core) according to standard procedures (see http://www.brightcore.be/). Raw data are quality controlled by use of FastQC (v0.10.1) and mapped to the human reference genome with BWA 0.7.10. Mapping qualities are assessed via overall coverage analysis by an in-house designed script. The mapped reads are processed using the GATK 2.7.2 (Genome Analysis Toolkit) pipeline (IndelRealaginer, BaseRecalibrator, HaplotypeCaller) and the detected variants are annotated by Annovar or Alamut Batch.
Patients 3, 4, and 7 were studied using targeted panel sequencing with Single-Molecule Molecular Inversion Probes as previously described (PMID: 27773430) [24]. For patients 5 and 6 whole-exome sequencing (WES) was done at the Broad Institute Genomic Services using Agilent SureSelect enrichment kit with subsequent sequencing of the libraries on a HiSeq 2000 (Illumina, San Diego, CA, USA). Sequence reads were aligned to the human genome (hg19) using BWA software or the CLC Biomedical Genomics workbench. Downstream processing was done with the Genome Analysis Toolkit, SAMtools, and Picard Tools. Single-nucleotide variants and indels were subsequently called by GATK Unified Genotyper (PMID:21478889) [25] and a variant quality score of ≥10 and were annotated using SeattleSeq SNP annotation and Annovar. Variants were then filtered using standard hard-filtering parameters (PMID:21478889) [25]. Specifically, only variants with a quality score of ≥30, sequencing depth of ≥10, quality/depth ratio of ≥5, length of homopolymer run of ≤5.0, and allelic balance of ≤0.80 were considered for downstream analysis.
For patient 8, WES was performed at the French National Centre for Genotyping (Evry, France). Library preparation, exome enrichment, WES, and analysis of variants were performed as previously described [26]. Exome sequencing quality data were homogeneous with an average mean depth higher than 100×. Coverage depth greater than 15× and 5× was obtained for about 97 and 99% of the target, respectively. We analyzed variants affecting coding regions and essential splice sites and excluded all variants with frequencies higher than 1% in multiple genome databases including the Single-Nucleotide Polymorphism Database, 1000 Genomes, the National Heart, Lung, and Blood Institute Exome Variant Server, the Exome Aggregation Consortium (ExAC), and a local Paris Descartes Bioinformatics platform database. The c.776C>T p.(Ser259Leu) variant was confirmed by Sanger sequencing and shown to be de novo. The reference sequence used was NM_001070.4 with systematic numbering of the exons (1–11); or as described in NG_033886.1. All variants have been submitted to https://databases.lovd.nl/shared/genes/TUBG1 (patient ID 00134040-00134047).
Results
We report four novel heterozygous variants in TUBG1 in eight patients, including two siblings. Clinical and imaging findings are summarized in Table 1.
At last examination patients were aged 18 months to 33 years. They all had moderate to severe intellectual disability, including very limited or absent language development, and variable degrees of motor impairment ranging from delayed motor development to severe spastic quadriplegia. Head circumference was available for five patients, one of whom was normocephalic and four were microcephalic (<−2.5SD). None of the patients showed dysmorphic facial features; one had congenital strabismus. Except for patient 3, all patients presented with seizures with a variable time of onset between the first months of life till after the 3rd year of life.
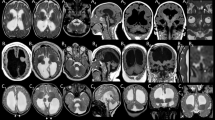
Brain magnetic resonance imaging (MRI) studies were performed between ages 2 month and 36 years (Figs. 1, 2). The cerebral cortex was most abnormal in patient 2 who had diffuse agyria. The other seven patients, five of which harbored the same c.776C>T variant, had a relatively homogeneous phenotype on imaging studies, characterized by diffuse pachygyria with a posterior to anterior gradient. The white matter showed a variable reduction in volume in six patients. Basal ganglia were dysplastic in patients 2 and 8. Brainstem and cerebellum were normal in all except for patient 2, who had brainstem and cerebellar vermis hypoplasia.

Imaging characteristics patients 1–4 and a normal subject. Brain MRI of patient 1 at age 36 years. Sagittal planes through the midline (a) show a normal corpus callosum, brainstem, and cerebellum. Axial T1-weighed images (b) and T2-weighed images (c) show pachygyria with a posterior to anterior gradient, enlarged posterior horns of the lateral ventricles, and enlarged perivascular spaces. Brain MRI of patient 2 at age 11 years. Sagittal planes (d) show a thin corpus callosum, hypoplasia of the brainstem, and the cerebellar vermis. Axial T2-weighed images (e, f) show diffuse lissencephaly, reduced white matter, and enlarged lateral ventricles and dysplastic basal ganglia. Brain MRI of patient 3 at age 1 year 6 months (g, h) and patient 4 at age 12 months (i, j, k). Sagittal planes (g, i) show a normal appearance of corpus callosum, brainstem, and the cerebellum. Axial T1-weighed images (h, j, k) show pachygyria with a posterior to anterior gradient, enlarged lateral ventricles, and reduced white matter. Brain MRI of a healthy subject (age 4 years). Sagittal image (l). Axial T2-weighed images (m, n)

Imaging characteristics patients 5–8. Brain MRI of patient 5 at age 13 years. Sagittal planes through the midline (a) show the absence of malformations of the corpus callosum, brainstem, and cerebellum. Axial T1-weighed images (b) and T2-weighed images (c) show pachygyria with a posterior to anterior gradient and enlarged posterior horns of the lateral ventricles. Brain MRI of patient 6 at age 2 months. Sagittal planes (d) show a thin corpus callosum, absence of hypoplasia of the brainstem, or the cerebellar vermis. Axial T2-weighed images (e, f) show diffuse pachygyria with a posterior to anterior gradient and enlarged lateral ventricles. Brain MRI of patient 7 at age 1 year 6 years. Sagittal images (g) show a normal appearance of corpus callosum, brainstem, and the cerebellum. Axial T2-weighed images (h, i) show pachygyria with a posterior to anterior gradient with almost normal frontal lobes, enlarged posterior horns of the lateral ventricles, and reduced white matter. Brain MRI of patient 8 at age 9 years. Sagittal images (j) show hypoplasia of the corpus callosum. Axial T2-weighed images (k, l) show pachygyria with a posterior to anterior gradient and important involvement of the temporal lobes, bilateral parietal infoldings, dysplastic basal ganglia, enlarged lateral ventricles, and reduced white matter
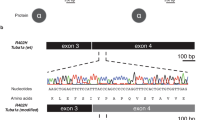
NGS analysis identified four novel heterozygous missense variants in TUBG1 (Table 2). Patients 3, 4, 5, 6, and 8 all share the same c.776C>T, p.(Ser259Leu) variant. In the siblings (patients 5 and 6), the variant was inherited by one of the parents who carries a germline mosaicism of this variant. Patient 7 is heterozygous for the c.769A>T, p.(Ile257Phe) variant, which is located very close to the recurrent c.776C>T variant. Currently, the c.776C>T variant has not been described in any database. Prediction programs SIFT, MutationTaster, PolyPhen2 all predict that this highly conserved nucleotide/amino acid is deleterious (Table 2). This variant is located in the Tubulin/FtsZ 2-layer sandwich (or C-terminal) domain (Fig. 3).

Distribution of the variants in the TUBG1 gene. Linear (a) and 3D (b) representation of the TUBG1 gene showing its functional domains and the distribution of the described TUBG1 variants. The variant in red in a represents the recurrent variant detected in four patients. The 3D structure is based on PDB ID: 3CB2 (crystal structure of gamma-tubulin bound to GDP) using NGL viewer [38, 39]
Discussion
Tubulinopathies have been characterized by a broad range of cortical malformations associated with hypoplasia or agenesis of the corpus callosum, dysmorphic basal ganglia, and hypoplasia of the brainstem, cerebellar vermis, and/or hemispheres [6, 9, 11, 27]. In some patients, the cerebellar dysgenesis and/or dysplastic basal ganglia may be more prominent than the cortical malformations [28].
Poirier et al. [14] initially described the phenotype associated with variants in TUBG1 as similar to that associated with variants in LIS1 (a.k.a. PAFAH1B1); an observation that has recently been integrated in the classification of lissencephalies proposed by DiDonato et al. [11]. A later report on the same patients mentioned that the two individuals with microcephaly and severe pachygyria resembled individuals with lissencephaly carrying the p.(Arg402Cys) substitution in TUBA1A [6].
In the current series including eight additional patients with variants in TUBG1, the most common imaging phenotype consists of partial or diffuse pachygyria with a posterior to anterior gradient, similar to the phenotype associated with variants in LIS1, DYNC1H1, or KIF5. However, the most severe end of the TUBG1-related spectrum also includes diffuse agyria as illustrated by patient 2, similar to the phenotype associated with some variants in LIS1 or the c.1205G>A, p.(Arg402His) variant in TUBA1A. The cortical malformation in patients with variants in TUBG1 therefore clearly stands out from that of tubulin-related dysgyria, which can be seen in patients with variants in TUBB2B, TUBB3, TUBB, and most variants in TUBA1A [11, 29].
In this study, associated brain abnormalities appear to be less frequent compared to other tubulinopathies. Except for two patients, all patients described so far had normal basal ganglia, which is usually considered a key feature for tubulinopathies and has been observed in 75% of patients [28]. Additionally, the brainstem and cerebellum were spared in most patients with TUBG1 variants, and if malformations in either of these two structures were present, they were usually subtle. This is in contrast to the high prevalence of 78.7% of cerebellar hypoplasia in individuals with variants in other tubulin genes [6, 28]. Polymicrogyria or polymicrogyria-like cortical dysplasia and a simplified gyral pattern, which is common in TUBB2B and TUBB3 variants, have not been described in patients with TUBG1 variants so far. The observation that TUBG1 causes undermigration leading to pachygyria or agyria can possibly be explained by a negative impact on cell morphology in patients carrying a TUBG1 variant, while variants in other tubulin genes are more often associated with dysgyria or overmigration presenting as polymicrogyria which suggests defective radial glial guidance of immature neurons [8].
Thus, the observation that the imaging phenotype associated with variants in TUBG1 differs from that associated with variants in other tubulin genes is in line with the function of TUBG1 in nervous system development and its stronger involvement in neuronal migration than in, for example, axon growth and orientation, which appears to be more prominently affected by variants in TUBB3 [21, 30, 31]. This hypothesis is also supported by the disturbed neuronal migration observed in an in utero RNA interference assay by Poirier et al. [14]. Most patients with variants in TUBG1 also exhibit microcephaly, indicating a major role of TUBG1 in neuronal cell proliferation.
It has been shown that tubulin isotypes have individual functions, expression levels, and distribution among different cell types, which led to the assumption that subtle phenotypic differences could exist. As the exact mechanism and the extent of how a variant alters the formation of functional tubulin heterodimers, GTP binding, longitudinal and lateral protofilament interactions, and microtubule interactions with microtubule-associated proteins remains widely unknown, so far, few conclusions can be drawn about these distinctive features [15, 21, 32, 33]. Nevertheless, the differences in physiological function of the TUBG1 protein as a scaffold in the formation of microtubules on the one hand and alpha-tubulin and beta-tubulin dimers as components of the microtubule on the other hand have been well established, and can give a possible explanation for the different phenotypic presentation on imaging [15].
The correlation between phenotype and genotype could further be determined by the exact location of the variant within the gene. This has been observed in the case of the recurrent c.1205G>A, p.(Arg402His) variant in TUBA1A causing classic lissencephaly, and the c.790C>T, p.(Arg264Cys) variant in the same gene associated with central pachygyria [6, 11, 34, 35]. Recurrent TUBB2B and TUBB3 variants have also been described to result in homogeneous phenotypes [15, 30, 36, 37]. This has also been observed in our study as the five patients with the c.776C>T, p.(Ser259Leu) variant shared a similar phenotype including mild frontal and moderate posterior pachygyria with no or subtle malformations of the corpus callosum, brainstem, and cerebellum. Additionally, patient 7 in our study carried a c.769A>T, p.(Ile257Phe) substitution which is only two amino acids upstream of the c.776C>T, p.(Ser259Leu) recurrent variant, and is associated with a similar phenotype. These variants are located at the borderline of the tubulin sandwich/C-terminal domain.
Interestingly, the c.985G>T, p.(Asp329Tyr) variant in patient 2, which was associated with the most severe phenotype in our series is located two amino acids upstream of the c.991A>C, p.(Thr331Pro) variant identified in a patient with posterior pachygyria and a moderate phenotype described by Poirier et al. [14]. These variants are also located in the tubulin sandwich/C-terminal domain. At this moment, it remains hard to predict the phenotype based on the position of the detected variants. However, c.985G>T, p.(Asp329Tyr) is located at the surface of the TUBG1 protein. In line with findings of the alanine-scanning mutagenesis, variants in surface proteins might have a more severe effect [22]. The majority of the variants are detected in the 2-layered sandwich domain of the TUBG1 protein, which is probably involved in the formation of dimers (Fig. 3). Consequently, these variants are expected to interfere with the microtubule formation and have a dominant-negative effect on the function of TUBG1.
So far, no truncating variants have been described in patients with TUBG1 variants. Whether truncations are likely to have either more severe phenotypes or no associated phenotype remains unclear. However, the number of variants described remains relatively small to draw conclusions.
Based on the current classification of lissencephalies, the majority of the patients with TUBG1 variants fit within subtype 2–3. The predicted clinical outcome based on the classification is concordant with the phenotype in our patients [11].
It is not yet possible to identify a particular causative tubulin gene or variant based on clinical and radiologic presentations alone, as findings are not specific enough. However, variants in TUBG1 should be considered as a possible differential diagnosis in patients presenting with posterior predominant pachygyria with no or minimal involvement of other brain structures, especially if variants in LIS1 have been ruled out.
References
Oakley CE, Oakley BR. Identification of gamma-tubulin, a new member of the tubulin superfamily encoded by mipA gene of Aspergillus nidulans. Nature. 1989;338:662–4.
Wise DO, Krahe R, Oakley BR. The gamma-tubulin gene family in humans. Genomics. 2000;67:164–70.
Rommens JM, Durocher F, McArthur J, et al. Generation of a transcription map at the HSD17B locus centromeric to BRCA1 at 17q21. Genomics. 1995;28:530–42.
Friedmann M, Holth LT, Zoghbi HY, Reeves R. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993;21:4259–67.
Draberova E, Sulimenko V, Vinopal S, et al. Differential expression of human gamma-tubulin isotypes during neuronal development and oxidative stress points to a gamma-tubulin-2 prosurvival function. FASEB J. 2017;31:1828–46.
Bahi-Buisson N, Poirier K, Fourniol F, et al. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain. 2014;137:1676–1700.
Breuss M, Heng JI, Poirier K, et al. Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2:1554–62.
Chakraborti S, Natarajan K, Curiel J, Janke C, Liu J. The emerging role of the tubulin code: from the tubulin molecule to neuronal function and disease. Cytoskeleton. 2016;73:521–50.
Cushion TD, Dobyns WB, Mullins JG, et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain. 2013;136:536–48.
Cushion TD, Paciorkowski AR, Pilz DT, et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am J Hum Genet. 2014;94:634–41.
Di Donato N, Chiari S, Mirzaa GM, et al. Lissencephaly: expanded imaging and clinical classification. Am J Med Genet A. 2017;173:1473–88.
Fallet-Bianco C, Laquerriere A, Poirier K, et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol Commun. 2014;2:69.
Poirier K, Keays DA, Francis F, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum Mutat. 2007;28:1055–64.
Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45:639–47.
Tischfield MA, Engle EC. Distinct alpha- and beta-tubulin isotypes are required for the positioning, differentiation and survival of neurons: new support for the ‘multi-tubulin’ hypothesis. Biosci Rep. 2010;30:319–30.
Mahoney NM, Goshima G, Douglass AD, Vale RD. Making microtubules and mitotic spindles in cells without functional centrosomes. Curr Biol. 2006;16:564–9.
Suri C, Hendrickson TW, Joshi HC, Naik PK. Molecular insight into γ-γ tubulin lateral interactions within the γ-tubulin ring complex (γ-TuRC). J Comput Aided Mol Des. 2014;28:961–72.
Raynaud-Messina B, Merdes A. Gamma-tubulin complexes and microtubule organization. Curr Opin Cell Biol. 2007;19:24–30.
Zheng Y, Wong ML, Alberts B, Mitchison T. Nucleation of microtubule assembly by a gamma-tubulin-containing ring complex. Nature. 1995;378:578–83.
Breuss MW, Leca I, Gstrein T, Hansen AH, Keays DA. Tubulins and brain development—the origins of functional specification. Mol Cell Neurosci. 2017;84:58–67. https://doi.org/10.1016/j.mcn.2017.03.002.
Tischfield MA, Cederquist GY, Gupta ML, Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev. 2011;21:286–94.
Hendrickson TW, Yao J, Bhadury S, Corbett AH, Joshi HC. Conditional mutations in gamma-tubulin reveal its involvement in chromosome segregation and cytokinesis. Mol Biol Cell. 2001;12:2469–81.
Aldaz H, Rice LM, Stearns T, Agard DA. Insights into microtubule nucleation from the crystal structure of human gamma-tubulin. Nature. 2005;435:523–7.
Di Donato N, Jean YY, Maga AM, et al. Mutations in CRADD result in reduced caspase-2-mediated neuronal apoptosis and cause megalencephaly with a rare lissencephaly variant. Am J Hum Genet. 2016;99:1117–29.
DePristo M, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Mirzaa GM, Conti V, Timms AE, et al. Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol. 2015;14:1182–95.
Romaniello R, Arrigoni F, Cavallini A, et al. Brain malformations and mutations in α- and β-tubulin genes: a review of the literature and description of two new cases. Dev Med Child Neurol. 2014;56:354–60.
Oegema R, Cushion TD, Phelps IG, et al. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum Mol Genet. 2015;24:5313–25.
Mutch CA, Poduri A, Sahin M, Barry B, Walsh CA, Barkovich AJ. Disorders of microtubule function in neurons: imaging correlates. AJNR Am J Neuroradiol. 2016;37:528–35.
Chew S, Balasubramanian R, Chan WM, et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013;136:522–35.
Whitman MC, Andrews C, Chan WM, et al. Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am J Med Genet A. 2016;170A:297–305.
Panda D, Miller HP, Banerjee A, Ludueña RF, Wilson L. Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc Natl Acad Sci USA. 1994;91:11358–62.
Leandro-García LJ, Leskelä S, Landa I, et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton. 2010;67:214–23.
Kumar RA, Pilz DT, Babatz TD, et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19:2817–27.
Mokánszki A, Körhegyi I, Szabó N, et al. Lissencephaly and band heterotopia: LIS1, TUBA1A, and DCX mutations in Hungary. J Child Neurol. 2012;27:1534–40.
Guerrini R, Mei D, Cordelli DM, Pucatti D, Franzoni E, Parrini E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet. 2012;20:995–8.
Amrom D, Tanyalçin I, Verhelst H, et al. Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin Genet. 2014;85:178–83.
Rose AS, Hildebrand PW. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015;43:W576–9.
Heinz DW, Baase WA, Dahlquist FW, Matthews BW. How amino-acid insertions are allowed in an alpha-helix of T4 lysozyme. Nature. 1993;361:561–4.
Acknowledgements
We thank the patients and their families for participation in this study.
Funding
AJ was supported by a Senior Clinical Investigator Fellowship from the Research Foundation Flanders (FWO) and received funding from the Scientific Fund Willy Gepts and the VUB Wetenschappelijk Steunfonds. NDD was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, DI 2079/2-1 and DI 2170/2-2). WBD was funded by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health under award numbers P01NS039404, R01NS050375, 1R01NS058721, and 1R01NS092772.
Author contributions
WBD, NDD, KS, and AJ conceived the work. SB, ES, MD, KK, and RG assisted with data acquisition. SB, KS, NDD, RG, and AJ drafted the manuscript, which was revised and approved by all.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: Stefanie Brock, Katrien Stouffs, Nataliya Di Donato, Anna C. Jansen.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brock, S., Stouffs, K., Scalais, E. et al. Tubulinopathies continued: refining the phenotypic spectrum associated with variants in TUBG1. Eur J Hum Genet 26, 1132–1142 (2018). https://doi.org/10.1038/s41431-018-0146-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0146-y
This article is cited by
-
Case reports: novel TUBG1 mutations with milder neurodevelopmental presentations
BMC Medical Genetics (2019)
-
TUBG1 missense variants underlying cortical malformations disrupt neuronal locomotion and microtubule dynamics but not neurogenesis
Nature Communications (2019)
