
Abstract
Acrodysostosis (MIM 101800) is a dominantly inherited condition associating (1) skeletal features (short stature, facial dysostosis, and brachydactyly with cone-shaped epiphyses), (2) resistance to hormones and (3) possible intellectual disability. Acroscyphodysplasia (MIM 250215) is characterized by growth retardation, brachydactyly, and knee epiphyses embedded in cup-shaped metaphyses. We and others have identified PDE4D or PRKAR1A variants in acrodysostosis; PDE4D variants have been reported in three cases of acroscyphodysplasia. Our study aimed at reviewing the clinical and molecular findings in a cohort of 27 acrodysostosis and 5 acroscyphodysplasia cases. Among the acrodysostosis cases, we identified 9 heterozygous de novo PRKAR1A variants and 11 heterozygous PDE4D variants. The 7 patients without variants presented with symptoms of acrodysostosis (brachydactyly and cone-shaped epiphyses), but none had the characteristic facial dysostosis. In the acroscyphodysplasia cases, we identified 2 PDE4D variants. For 2 of the 3 negative cases, medical records revealed early severe infection, which has been described in some reports of acroscyphodysplasia. Subdividing our series of acrodysostosis based on the disease-causing gene, we confirmed genotype–phenotype correlations. Hormone resistance was consistently observed in patients carrying PRKAR1A variants, whereas no hormone resistance was observed in 9 patients with PDE4D variants. All patients with PDE4D variants shared characteristic facial features (midface hypoplasia with nasal hypoplasia) and some degree of intellectual disability. Our findings of PDE4D variants in two cases of acroscyphodysplasia support that PDE4D may be responsible for this severe skeletal dysplasia. We eventually emphasize the importance of some specific assessments in the long-term follow up, including cardiovascular and thromboembolic risk factors.
Similar content being viewed by others


Introduction
Acrodysostosis is a rare autosomal dominant condition first described by Pierre Maroteaux et al. in 1968 [1] and further reviewed by Robinow et al. in 1971 [2]. It consists in the association of (1) skeletal features characterized by short stature, facial dysostosis with nasal hypoplasia and peripheral dysostosis with severe brachymetatarsia, brachymetacarpia, brachydactyly, cone-shaped epiphyses, and advanced bone maturation, (2) inconstant resistance to multiple hormones including parathyroid hormone or thyrotropin, and (3) possible neurological involvement with moderate to mild intellectual disability [1, 2]. Differential diagnoses include Albright hereditary osteodystrophy (AHO) [MIM103580] and pseudopseudohypoparathyroidism [MIM 612463] due to loss of function variants in GNAS (α-stimulary subunit of the G-protein) and characterized by less severe skeletal involvement [3].
In 2011, the recurrent p.Arg368* variant in the PRKAR1A gene has been found in three individuals with acrodysostosis and resistance to multiple hormones [4]. PRKAR1A encodes the cyclic AMP (cAMP)-dependent regulatory subunit of protein kinase A. The mutated subunit impairs the protein kinase A response to cAMP [4], accounting for hormone resistance and skeletal abnormalities resembling those observed in AHO. We then identified PDE4D variants as another cause of acrodysostosis, most frequently without hormone resistance [5]. PDE4D is also involved in cAMP signaling pathway metabolism, encoding a class IV cAMP-specific phosphodiesterase, regulating cAMP concentration.
On the basis of these molecular basis, two distinct MIM references have been generated, namely ACRO1 (MIM 101800) for PRKAR1A variants and ACRO2 (MIM614613) for PDE4D variants. We previously reported some genotype–phenotype correlations. Indeed, patients with PRKAR1A variants tend to have less characteristic facial dysostosis, normal intelligence, and resistance to multiple hormones compared to patients with PDE4D variants who presented with characteristic facial dysostosis, intellectual disability, and subtle or absence of hormonal resistance [6, 7].
In recent studies, several acroscyphodysplasia cases [MIM 250215] [7, 8] were reported with a PDE4D variant. This entity consists of the association of severe growth retardation, micromelia predominating in the lower limbs, knee flexion, and severe brachydactyly altogether with a specific radiological appearance of the knees: cup-shaped metaphyses with embedded epiphyses [9]. This radiological aspect is reminiscent of the appearance of the cone-shaped epiphyses of small joints in acrodysostosis.
Studying 32 unrelated cases of acrodysostosis (n = 27) or acroscyphodysplasia (n = 5), we found PDE4D variants in 13 cases, including 2 cases of acroscyphodysplasia, and PRKRA1A variants in 9 acrodysostosis cases. We confirmed some genotype–phenotype correlations and involvement of PDE4D in acroscyphodysplasia. We also emphasize the importance of some specific assessments in the long-term follow up of these conditions.
Subjects and methods
Clinical ascertainment
Overall, 32 patients were recruited for this study, including 27 cases of acrodysostosis (comprising 10 patients previously described in Michot et al. [5]) and 5 cases of acroscyphodysplasia. Two patients had an affected parent. Inclusion criteria for acrodysostosis cases were a peripheral dysostosis with severe generalized brachydactyly, affecting metacarpals and phalanges, associated with cone-shaped epiphyses. Short stature, facial dysostosis, resistance to multiple hormones, and intellectual disability were not considered as mandatory. The acroscyphodysplasia cases were recruited on the sole criterion of a characteristic radiological aspect of the knees.
Among the 32 recruited patients, 18 were females and 14 were males. Thirty patients were sporadic cases, and in 2 cases, acrodysostosis was inherited from an affected mother.
Informed consent for participation, sample collection, and photograph publication were obtained using protocols approved by the Necker Hospital ethics board committee.
Clinical data collection
According to a literature review, a questionnaire with selected medical items was sent out to the referring physician, to collect details on the clinical and biological symptoms of the cases diagnosed with acrodysostosis and acroscyphodysplasia. Available data, photographs, and radiographs (when authorized by the patients or their legal representatives) were collected.
Hormonal screening
For complete screening of mineral metabolism, blood levels of creatinine, calcium, phosphorus, thyroxin (T4), thyrotropin, 25-hydroxyvitamin D, 1-25-dihydroxyvitamin D, parathyroid hormone (PTH), and fibroblast growth factor 23 (FGF23) levels were measured, along with urinary creatinine, calcium, and phosphorus excretion.
PRKAR1A and PDE4D molecular screening
Genomic DNA was obtained from peripheral blood leukocytes using standard procedures. The exons and exon–intron boundaries of PRKAR1A (GenBank NM_ 002734.4) and PDE4D (GenBank NM_001104631.1) were amplified using specific primers (available upon request). Amplification products were purified by ExoSapIT (Amersham, Buckinghamshire, UK) and directly sequenced using the Big Dye Terminator Cycle Sequencing Ready Reaction kit v.1.1 on an automatic sequencer (ABI3130xl; PE Applied Biosystems, Foster City, CA). Sequence analyses were performed using Seqscape software v2.5 (Applied Biosystems).
Results
Sequencing data
De novo heterozygous PRKAR1A variants were identified in 9 cases, including the p.Arg368* stop variant [4] in 5/9 and missense variants (c.786G>C (p.(Trp262Cys)), c.968A>C (p.(Tyr323Ser)), c.968A>G (p.(Tyr323Cys)), c.1117T>C (p.(Tyr373His))) in 4/9. All these variants were predicted as probably damaging using PolyPhen2 software and were not identified in 200 control chromosomes, nor indexed in gnomAD database. They altered a conserved amino acid, located either in the catalytic domain or in the cAMP-binding domain B (Fig. 1).

Scheme of the PRKAR1A gene, modified from Linglart el al. [7]. Above are listed the previously published variants, including the recurrent p.Arg368* variant, which was found in four cases in this study. Below are the four new and unique variants described in this study
PDE4D variants were identified in 11 acrodysostosis cases, including the two familial cases. They were all missense variants spread throughout the whole PDE4D coding sequence (Fig. 2), altering a conserved amino acid located in the catalytic domain and were predicted as probably damaging using PolyPhen2 software, except for the c.568T>G (p.(Ser190Ala)) and c.568T>C (p.(Ser190Pro)) predicted as benign. These last 2 variants occurred de novo and affected a serine residue predicted to be phosphorylated (UniProt database) and were absent from alleles in 200 ethnicity-matched controls and from gnomAD database.

Scheme of the PDE4D gene. Above are listed the previously published variants. Below are the eleven new and unique variants described in this study
De novo PDE4D variants (c.995T>C (p.(Phe332Ser)) and c.99T>G (p.(Ile333Met))) were identified in 2/5 patients with acroscyphodysplasia.
Clinical, biological, and radiological data of acrodysostosis cases
The clinical and available biological details are summarized in Table 1 for patients with identified variants.
Intrauterine growth retardation (IUGR) was observed in 8/9 with PRKAR1A variants, but only in a few patients with PDE4D variants (n = 5/11, but no available data in three cases). Postnatally, 8/9 patients with PRKAR1A variants developed growth deficiency, ranging from −2 SD to −6.7 SD, whereas only 4/11 individuals with PDE4D variants had postnatal short stature. However, patients with PDE4D variants reported herein were young at time of the study (6 were below 10 years of age). Data on pubertal development were not available for these young patients, but one boy had cryptorchidism treated by testosterone and one girl a female hypospadias. One boy with PRKAR1A variant (out of 3) also had cryptorchidism, which required surgery. In four adult women with PRKAR1A variants, one experienced a miscarriage and an intrauterine fetal death, whereas another one had an absence of the mammary glands development.
Brachydactyly involved all rays of fingers and toes, associated with cone-shaped epiphyses. Three patients also had single palmar crease, including two with PRKAR1A variants and one without any identified variant. A relative hyperplasia of the first ray of the feet was historically described and was indeed observed in 6/9 patients with PRKAR1A variants and 2/11 with PDE4D variants. One patient with PDE4D variant (patient 20) developed a macrodactyly of second and third rays of one foot that required amputation. Elder patients with PRKAR1A variants developed stiffness of the elbows and one underwent corrective surgery for carpal tunnel syndrome.
A flat face with malar hypoplasia was noted in all patients (Fig. 3). Nevertheless the distinctive nasal hypoplasia was described in only 3/9 patients with PRKAR1A variants, whereas 11/11 patients with PDE4D variants had this typical facial feature. Noteworthy, the three patients with PRKAR1A variants and nasal hypoplasia harbored missense variants affecting the Tyrosine 323 (c.786G>C, c.968A>C and c.968A>G) and the tryptophane 262 residues, and not the recurrent stop variant affecting arginine 368 residue. Moreover, 3/9 patients with PRKAR1A variants and 5/11 with PDE4D had a prominent mandible. Two patients with PRKAR1A variants presented with delayed eruption of teeth and 1 with PDE4D variant had abnormal enamel.

Patients presenting with acrodysostosis. a, b Patient 5; general aspect and hand X-ray. c–e Patient 9; face, hand, and hand X-ray. f–h Patient 15; face, hand, and hand X-ray. Please note on c and f the typical facial gestalt with malar hypoplasia and nasal hypoplasia. Please note on b, e, and h the short broad metacarpals and phalanges, with cone-shaped epiphyses for patient 15
Eight out of nine with PRKAR1A variant had normal intellectual development. Only patient 8 (who harbored a PRKAR1A variant affecting the tyrosine 323 residue (c.968A>C)) had a moderate intellectual disability with congenital axial hypotonia and triventricular cerebral dilatation. Noteworthy, the second patient with a variant affecting this residue, patient 9 (c.968A>G), was only 18 months old at the time of completion and besides he had walked normally at 15 months of age, congenital hypotonia and poor sucking had been noticed before. Conversely almost all patients (10/11) with PDE4D variants had mild to moderate intellectual disability. Moreover, two patients with a PDE4D variant developed acute intracranial hypertension due to a thrombophlebitis (patients 11 and 12).
All patients with PRKAR1A variants had endocrine disorders: 8/9 had an increased level of parathyroid hormone (PTH), 7/9 presented with peripheral hypothyroidism, and 2/9 with growth hormone deficiency. Conversely, only 1 patient with PDE4D variant had slightly increased level of PTH and no patient experienced hypothyroidism.
Additional health issues were observed including (i) recurrent ENT infections (n = 2; both with PRKAR1A variants), (ii) chronic erysipelas with fixed edema of the lower limbs and hypogammaglobulinemia (n = 1; PRKAR1A variant), and (iii) deafness (n = 3; two PRKAR1A variants and one with PDE4D).
Moreover, both patients with PRKAR1A variant affecting the tyrosine 323 residue (c.968A>C and c.968A>G) had additional symptoms including cardiovascular malformations (one with external jugular vein stenosis and one with total anomalous pulmonary venous return with atrial septal defect). One (patient 8 (c.968A>C)) had severe laryngotracheomalacy with cricoid hemangioma, whereas the other (patient 9 (c.968A>G)) required the dilatation of a subglottic diaphragm in infancy.
Noteworthy, one of the elder patients (patient 5) with the p.Arg368* PRKAR1A variant died suddenly at 38 years of age, following severe bronchospasm during a surgical procedure. She had concomitant lactic acidosis, rhabdomyolysis, and anuric renal deficiency. No clear etiology was determined despite extensive metabolic and infectious work-up.
All patients had shortened tubular bones of the hands and feet, of all the digits, associated with cone-shaped epiphyses (Fig. 3). Besides these canonical signs, other radiological features were noticed: advanced bone age (in 4/9 with PRKAR1A variants and 6/11 with PDE4D), bilateral coxa valga (4/9 with PRKAR1A variants and 3/11 with PDE4D), irregular vertebral plates (2/9 with PRKAR1A variants and 2/11 with PDE4D), absence of widening of the interpedicular distance (3/9 with PRKAR1A variants and 2/11 with PDE4D). Moreover, 5/9 patients with PRKAR1A variants had skeletal abnormalities also affecting other epiphyses and one case had an important vertebral dysplasia in the first years of life, which progressively improved with age.
Clinical, biological, and radiological data of the five acroscyphodysplasia cases
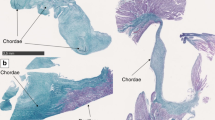
All patients but one presented with postnatal growth retardation (−2 to −4 SD), after intrauterine growth retardation in only one case. Four patients have nasal hypoplasia, severe in two cases only; all five had a flat face with malar hypoplasia. They all had also micromelia and 3/5 had a severe brachydactyly with small phalanges and small metacarpals and metatarsals (two with moderate brachydactyly) (Fig. 4). Cone-shaped epiphyses were observed except in one young case. Two patients had knee stiffness. One patient presented with a peripheral hypothyroidism, but no other endocrine disorder was reported. All had an intellectual disability. Some non-specific MRI abnormalities were observed (one with thin corpus callosum, delayed myelinisation and cortical atrophy, another one with ventricular dilatation).

X-rays of the acroscyphodysplasia cases showing the whole legs, the zoom on the knee and a hand. a–c 1st case. d–f 2nd case. g–i 3rd case. j–l 4th case. m–o 5th case. Please note the cup-shaped metaphyses with embedded epiphyses on knees and the aspect of the hands, similar to the acrodysostosis cases
Among the 3 patients with no identified variants, 2 had a medical history of an early and severe infection and the last one of frequent infections.
Discussion
We describe here a large cohort of 27 patients with acrodysostosis. We identified variants in 74% of cases (20/ 27) including PDE4D variants in 11 and PRKAR1A in 9.
We further confirm genotype–phenotype correlations. Indeed, the 11 patients carrying PDE4D variants had the characteristic facial features with nasal hypoplasia, midface hypoplasia, and prominent mandible initially reported in acrodysostosis, and 10/11 had some degree of intellectual disability. This characteristic facial dysostosis and mild to moderate intellectual disability was observed, respectively, in only 3/9 and 1/9 cases carrying PRKAR1A variant. Noteworthy those patients had variants affecting the tryptophane 262 or the tyrosine 323 residues of PRKAR1A (c.786G>C, c.968A>C, and c.968A>G), but not the recurrent p.Arg368* variant. Conversely, hormone resistance was consistently observed in patients carrying PRKAR1A variants (8/9 presenting with chronic resistance to parathyroid hormone and 7/9 had peripheral hypothyroidism), whereas only one case with PDE4D variant had a slightly increased level of PTH.
These results are in agreement with all cases reported so far (n = 49; 24 with PRKAR1A variants and 25 with PDE4D variants) [4, 6, 7, 10,11,12,13,14,15,16] (cf Table 2). In the literature, the peripheral bone dysostosis is consistently observed in all patients. The severe brachymetacarpia/brachymetatarsia and brachydactyly affect all the digits. The lumbar stenosis is also described in patients with PRKAR1A or PDE4D variants, but this data is scarcely specified in literature. Although short nose with flat face is consistently observed in all reported cases, severe nasomaxillary hypoplasia is more frequently reported in patients with PDE4D variants (n = 24 for whom data are specified), than in patients with PRKAR1A variants (n = 6 for whom data are specified). Intellectual disability is clearly reported in 24 cases of patients with PDE4D variants (of 25 reported in literature) but only in 5 with PRKAR1A variants (of 24 reported). Hormone resistance is conversely observed in only 6 patients carrying PDE4D variants (6/25 cases), whereas is present in 17 patients with PRKAR1A variants (of 24 cases (19 with the data available)) in literature.
We therefore confirm that molecular analysis shall be directed according to the symptoms: in case of facial dysostosis and moderate intellectual disability, first screen the PDE4D gene, and in case of less characteristic facial features and hormonal resistance first screen for PRKAR1A variants.
Our study also further highlights the risk of development of serious medical complications. Actually 3 patients with PRKAR1A and 2 with PDE4D variants developed progressive obesity which confirms previous publications [6]. cAMP signaling pathways, mediated by protein kinase A (PKA), have an important role in the metabolic homeostasis including the regulation of adiposity [17]. In mice, targeted disruption of the RII beta gene, coding for a regulatory subunit of PKA, leads to a lean phenotype with resistance to diet-induced obesity [18]. Cushing syndrome with bilateral adrenocortical hyperplasia may be caused by inactivating variants of PRKAR1A leading to an increase of PKA activation. The patients with these inactivating PRKAR1A variants have a lower BMI than the ones without variants, confirming a link between germline defects of PKA and human obesity phenotypes [19].
Similarly, PDE4D which converts cAMP to AMP, countering activation of PKA, is an insulin-responsive gene important for metabolism regulation, including adipocyte lipolysis [20]. Rat adipocytes treated with the PDE4 inhibitor rolipram showed a significant increase in lipolysis and reversed in part prostaglandin E2 antilipolysis [20]. Furthermore, cAMP-dependent PKA pathway is also involved in insulin secretion and resistance. Overexpression of PDE4D in α-cells reduced insulin secretion, whereas inhibition of PDE4 activity by rolipram or knockdown of PDE4D restored it [21]. One patient with acrodysostosis has clearly been described with a metabolic syndrome and hypertension [22]. Although the physiopathological link between PRKAR1A and PDE4D variants and human obesity and insulin resistance will require further study, follow-up of patients should be careful on the BMI, blood pressure, and glucose status.
We also report two patients with PDE4D variants who developed intracranial hypertension and thrombophlebitis. One adult patient with acrodysostosis and one child with PDE4D variant have also been described with a deep vein thrombosis [13, 23]. Actually, several case–control studies have assigned an association between PDE4D variants and risk of ischemic stroke among different ethnicities [24]. Moreover, PDE4D is associated with inflammation and reduced PDE4D is assumed to predispose individuals to atrial fibrillation, which increases the risk of cardiogenic stroke [25]. More studies are necessary to assess the functional and physiological effects of the described PDE4D SNPs. We advise to pay special attention to the thromboembolic risk during follow-up. Actually we emphasize the importance of a multidisciplinary long-term follow up, including cardiovascular and thromboembolic risk factors and we propose a short checklist for medical management (Table 3).
In this study, we also investigated 5 rare cases of acroscyphodysplasia initially described by Verloes et al. in 1991 [9]. All 5 patients had the characteristic metaphyseal changes, but only 3 patients had very short hands and feet, considered as a main criterion for this diagnosis [9, 26]. Among these 3 patients, PDE4D variants were identified in the 2 with severe growth retardation (−3 and −4 SD), severe nasal hypoplasia and stiffness of the knees. This finding suggests that PDE4D screening should be considered in case of knee cup-shaped metaphyses with embedded epiphyses when associated with acrodysostosis-like brachydactyly and nasal hypoplasia. As few observations of typical knee cup-shaped metaphyses with embedded epiphyses have been described after repeated injuries or meningococcemia [26], the reported infections could be at least a part of the cause of the aspect of acroscyphodysplasia in 2/3 patients without identified PDE4D variants. A recent study has described GNAS variants in 2 cases of acroscyphodysplasia, supporting the hypothesis of a genetic heterogeneity for this condition [8].
The majority of described cases of acrodysostosis have been sporadic with no familial medical history. In our study, we described two cases of mother-to-child transmission of PDE4D variants (patients 17 and 18). Prior to PRKAR1A and PDE4D identification, several reports have described autosomal dominant inheritance, from mother [27,28,29,30,31] or father [32]. Only one report described concordant siblings with seemingly unaffected parents [33], but it has been supported by the observation of Lynch et al. [6]. They described three affected children with clinically unaffected parents, but with a PDE4D variant-carrying father. They raise the hypothesis of an imprinting of PDE4D gene to explain the variable expressivity, as in the mouse genome, PDE4D showed a paternal bias in transmission [34]. However, the familial cases we described with a maternal transmission of PDE4D variants and clear phenotype in the kindred rather opposes this hypothesis. More familial cases will be needed to further accord the resulting phenotypes regarding the parental inheritance.
Finally, neither PDE4D nor PRKAR1A variants were found in seven patients with characteristic skeletal features but no hormone resistance or facial dysostosis, supporting that other disease genes may account for these remaining acrodysostosis cases. Interestingly, both forms of acrodysostosis (ACRO1—MIM 101800—corresponding to variants of PRKAR1A and ACRO2—MIM614613—to variants of PDE4D) are due to variants of two genes of the same pathway of cAMP signaling. PRKAR1A is the cAMP-dependent regulatory subunit of protein kinase A and PDE4D is a class IV cAMP-specific phosphodiesterase, regulating cAMP concentration. The involvement of PDE4D in acrodysostosis further supports the key role of cAMP signaling pathway in skeletogenesis, as previously shown for Albright hereditary osteodystrophy due to GNAS variants. The conditions share several clinical features (metacarpal abnormalities, obesity, and resistance to hormones depending on generation of cAMP for acrodysostosis with PRKAR1A variants), but some manifestations are variable in severity (degree and extent of brachydactyly and degree of parathyroid hormone resistance) and some others are quite similar. This clinical variability may be possibly linked to the tissue-specificity of the imprinting of GNAS or to the tissue-specific expression of alternative isoforms of protein kinase A or of phosphodiesterase 4 [4]. Finally, the dysregulation of cAMP intracellular signaling in response to a number of membrane-impermeable hormones could be the underlying common mechanism of acrodysostosis. Further study on the genomes of patients without identified variants is required to elucidate the molecular basis in these cases, but it might be hypothesized that the putative other genes involved in acrodysostosis are linked to regulation of cAMP signaling.
References
Maroteaux P, Malamut G. Acrodysostosis. Presse Med. 1968;76:2189–92.
Robinow M, Pfeiffer RA, Gorlin RJ, et al. Acrodysostosis. A syndrome of peripheral dysostosis, nasal hypoplasia, and mental retardation. Am J Dis Child. 1971;121:195–203.
Bastepe M, Juppner H. GNAS locus and pseudohypoparathyroidism. Horm Res. 2005;63:65–74.
Linglart A, Menguy C, Couvineau A, et al. Recurrent PRKAR1A variant in acrodysostosis with hormone resistance. N Engl J Med. 2011;364:2218–26.
Michot C, Le Goff C, Goldenberg A, et al. Exome sequencing identifies PDE4D variants as another cause of acrodysostosis. Am J Hum Genet. 2012;90:740–5.
Lynch DC, Dyment DA, Huang L, et al. Identification of novel variants confirms PDE4D as a major gene causing acrodysostosis. Hum Mutat. 2013;34:97–102.
Linglart A, Fryssira H, Hiort O, et al. PRKAR1A and PDE4D variants cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. 2012;97:E2328–2338.
Mitsui T, Kim OH, Hall CM, et al. Acroscyphodysplasia as a phenotypic variation of pseudohypoparathyroidism and acrodysostosis type 2. Am J Med Genet A. 2014;164A:2529–34.
Verloes A, Le Merrer M, Farriaux JP, Maroteaux P. Metaphyseal acroscyphodysplasia. Clin Genet. 1991;39:362–9.
Kaname T, Ki CS, Niikawa N, et al. Heterozygous variants in cyclic AMP phosphodiesterase-4D (PDE4D) and protein kinase A (PKA) provide new insights into the molecular pathology of acrodysostosis. Cell Signal. 2014;26:2446–59.
Lee H, Graham JM Jr., Rimoin DL, et al. Exome sequencing identifies PDE4D variants in acrodysostosis. Am J Hum Genet. 2012;90:746–51.
Li N, Nie M, Li M, et al. The first variant identified in a Chinese acrodysostosis patient confirms a p.G289E variation of PRKAR1A causes acrodysostosis. Int J Mol Sci. 2014;15:13267–74.
Lindstrand A, Grigelioniene G, Nilsson D, et al. Different variants in PDE4D associated with developmental disorders with mirror phenotypes. J Med Genet. 2014;51:45–54.
Muhn F, Klopocki E, Graul-Neumann L, et al. Novel variants of the PRKAR1A gene in patients with acrodysostosis. Clin Genet. 2013;84:531–8.
Nagasaki K, Iida T, Sato H, et al. PRKAR1A variant affecting cAMP-mediated G protein-coupled receptor signaling in a patient with acrodysostosis and hormone resistance. J Clin Endocrinol Metab. 2012;97:E1808–1813.
Rhayem Y, Le Stunff C, Abdel Khalek W, et al. Functional characterization of PRKAR1A variants reveals a unique molecular mechanism causing acrodysostosis but multiple mechanisms causing carney complex. J Biol Chem. 2015;290:27816–28.
McKnight GS, Cummings DE, Amieux PS, et al. Cyclic AMP, PKA, and the physiological regulation of adiposity. Recent Prog Horm Res. 1998;53:139–59.
Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–6.
London E, Rothenbuhler A, Lodish M, et al. Differences in adiposity in Cushing syndrome caused by PRKAR1A variants: clues for the role of cyclic AMP signaling in obesity and diagnostic implications. J Clin Endocrinol Metab. 2014;99:E303–10.
Wang H, Edens NK. mRNA expression and antilipolytic role of phosphodiesterase 4 in rat adipocytes in vitro. J Lipid Res. 2007;48:1099–107.
Kim MJ, Park SK, Lee JH, et al. Salt-inducible kinase 1 terminates cAMP signaling by an evolutionarily conserved negative-feedback loop in beta-cells. Diabetes. 2015;64:3189–202.
Atabek ME, Pirgon O, Sert A. Metabolic syndrome manifestations in an adolescent with acrodysostosis. J Pediatr Endocrinol Metab. 2007;20:739–41.
Sezer N, Sutbeyaz ST, Koseoglu F, Aras M, Akin C. Adult case of acrodysostosis with severe neurologic involvement. J Back Musculoskelet Rehabil. 2009;22:125–9.
Das S, Roy S, Munshi A. Association between PDE4D gene and ischemic stroke: recent advancements. Int J Neurosci. 2016;126:577–83.
Jorgensen C, Yasmeen S, Iversen HK, Kruuse C. Phosphodiesterase-4D (PDE4D)—a risk factor for atrial fibrillation and stroke? J Neurol Sci. 2015;359:266–74.
Dieux-Coeslier A, Moerman A, Holder M, et al. Metaphyseal chondrodysplasia with cone-shaped epiphyses: a specific form involving the lower limbs. Am J Med Genet A. 2004;124A:60–6.
Cantú JM, Hernández A, Panduro-Cerda A, et al. Autosomal dominant acrodysostosis. Hum Genet. 1979;47:345–6.
Hernández RM, Miranda A, Kofman-Alfaro S. Acrodysostosis in two generations: an autosomal dominant syndrome. Clin Genet. 1991;39:376–82.
Niikawa N, Matsuda I, Ohsawa T, Kajii T. Familial occurrence of a syndrome with mental retardation, nasal hypoplasia, peripheral dysostosis, and blue eyes in Japanese siblings. Hum Genet. 1978;42:227–32.
Sheela SR, Perti A, Thomas G. Acrodysostosis: autosomal dominant transmission. Indian Pediatr. 2005;42:822–6.
Steiner RD, Pagon RA. Autosomal dominant transmission of acrodysostosis. Clin Dysmorphol. 1992;1:201–6.
Davies SJ, Hughes HE. Familial acrodysostosis: can it be distinguished from Albright's hereditary osteodystrophy? Clin Dysmorphol. 1992;1:207–15.
Taillet-Bellemère C, Maroteaux P. Acrodysostosis in a sister and brother born to normal parents. Ann Pediatr. 1991;38:31–6.
Babak T, Deveale B, Armour C, Raymond C, Cleary MA, van der Kooy D, Johnson JM, Lim LP. Global survey of genomic imprinting by transcriptome sequencing. Curr Biol. 2008;18:1735–41.
Acknowledgements
We thank all patients and their families for their contribution to this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Michot, C., Le Goff, C., Blair, E. et al. Expanding the phenotypic spectrum of variants in PDE4D/PRKAR1A: from acrodysostosis to acroscyphodysplasia. Eur J Hum Genet 26, 1611–1622 (2018). https://doi.org/10.1038/s41431-018-0135-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0135-1
This article is cited by
-
Protein Kinase A in neurological disorders
Journal of Neurodevelopmental Disorders (2024)
-
Growth patterns and outcomes of growth hormone therapy in patients with acrodysostosis
Journal of Endocrinological Investigation (2023)
-
Ablation of specific long PDE4D isoforms increases neurite elongation and conveys protection against amyloid-β pathology
Cellular and Molecular Life Sciences (2023)
-
A novel variant in the PDE4D gene is the cause of Acrodysostosis type 2 in a Lithuanian patient: a case report
BMC Endocrine Disorders (2021)
-
Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders
Molecular Psychiatry (2021)
