
Abstract
Pontocerebellar hypoplasia (PCH) represents a group of autosomal-recessive progressive neurodegenerative disorders of prenatal onset. Eleven PCH subtypes are classified according to clinical, neuroimaging and genetic findings. Individuals with PCH type 9 (PCH9) have a unique combination of postnatal microcephaly, hypoplastic cerebellum and pons, and hypoplastic or absent corpus callosum. PCH9 is caused by biallelic variants in AMPD2 encoding adenosine monophosphate deaminase 2; however, a homozygous AMPD2 frameshift variant has recently been reported in two family members with spastic paraplegia type 63 (SPG63). We identified homozygous or compound heterozygous AMPD2 variants in eight PCH-affected individuals from six families. The eight variants likely affect function and comprise one frameshift, one nonsense and six missense variants; seven of which were novel. The main clinical manifestations in the eight new patients and 17 previously reported individuals with biallelic AMPD2 variants were postnatal microcephaly, severe global developmental delay, spasticity, and central visual impairment. Brain imaging data identified hypomyelination, hypoplasia of the cerebellum and pons, atrophy of the cerebral cortex, complete or partial agenesis of the corpus callosum and the “figure 8” shape of the hypoplastic midbrain as consistent features. We broaden the AMPD2-related clinical spectrum by describing one individual without microcephaly and absence of the characteristic “figure 8” shape of the midbrain. The existence of various AMPD2 isoforms with different functions possibly explains the variability in phenotypes associated with AMPD2 variants: variants leaving some of the isoforms intact may cause SPG63, while those affecting all isoforms may result in the severe and early-onset PCH9.
Similar content being viewed by others


Introduction
Pontocerebellar hypoplasia (PCH) represents a group of autosomal-recessive progressive neurodegenerative disorders of prenatal onset. Up to date, eleven PCH subtypes (PCH1-PCH11) have been identified according to clinical, neuroimaging and genetic findings [1,2,3,4,5]. In all subtypes, imaging and neuropathology show hypoplasia followed by variable atrophy of the cerebellum and pons. Clinical features include progressive microcephaly, severe developmental delay and additional neurologic symptoms depending on the subtype [3].
The combination of hypoplastic cerebellum and pons, hypoplastic or absent corpus callosum, and postnatal microcephaly is unique to pontocerebellar hypoplasia type 9 (PCH9; MIM 615809) [6]. In addition, a characteristic “figure 8” shape of the midbrain on axial brain images seems to represent a pathognomonic structural anomaly for this PCH type [6]. PCH9 is characterized by a severe course, with developmental delay, seizures, and spasticity. Biallelic variants affecting function in the gene encoding adenosine monophosphate deaminase 2 (AMPD2) have been reported to underlie PCH9 [6,7,8,9]. AMPD2 converts adenosine monophosphate to inosine monophosphate, thereby playing an evolutionary conserved role in maintaining cellular guanine pools [6, 10]. PCH9-associated variants in AMPD2 cause a severe reduction or even absence of AMPD2 protein that has been linked to adenosine-mediated neurotoxicity and defective GTP-dependent protein translation as the pathogenic mechanisms for this PCH type [6]. The AMPD2-associated phenotypic spectrum has been further expanded by the report of four families: one had a single male offspring exhibiting classical PCH9 [8], one had five siblings with PCH9-overlapping features in all and axonal peripheral neuropathy in two [7], one had three siblings with severe PCH9 in addition to peculiar craniofacial dysmorphism and teeth abnormalities [9], and the fourth family had two members exhibiting hereditary spastic paraplegia (HSP) and no PCH [SPG63; MIM 615686] [11]. Functional redundancy of AMPD2 isoforms and AMPD paralogs, such as AMPD1 and AMPD3, has been suggested to contribute to the variability of phenotypes caused by biallelic AMPD2 variants [7, 8].
We report eight individuals from six unrelated families with novel and known biallelic AMPD2 variants and evaluate the clinical and genetic data of these patients and the individuals reported previously. We also review the current knowledge on genotype-phenotype correlation in patients with AMPD2 variants and the pathogenic mechanisms underlying PCH.
Material and methods
Whole-exome sequencing and data analysis
Informed consent for DNA storage and genetic analyses was obtained from the parents/legal guardians of all subjects, and genetic studies were approved by all Institutional Review Boards of the participating institutions. For family 1, whole-exome sequencing (WES) was performed on genomic DNA extracted from leukocytes of patient 1 and two unaffected siblings. Enrichment was carried out using the Nextera® Enrichment Kit (62 Mb) (Illumina) according to the manufacturer’s protocols. Each captured library was then loaded and sequenced on the HiSeq 2500 platform (Illumina). Variant analysis was performed as previously described [12]. Briefly, the workflow of the Genome Analysis Toolkit (GATK) [13] recommended by the developers was applied. Afterwards, variants were functionally annotated and compared to those documented in publicly accessible genetic variant databases (dbSNP138, 1000 Genomes, and ExAC) using AnnoVar (v2015-03-22) [14]. Only exonic sequence alterations and intronic variants at exon–intron boundaries ranging from −40 to +40, neither present in the homozygous state in variant databases nor in the unaffected siblings, with unknown frequency or a minor allele frequency <0.5% were retained.
In family 2, WES was performed on the index patient (patient 2) only. Enrichment was performed with NimbleGen SeqCap EZ Human Exome Library v3.0 (Roche) and sequencing on a HiSeq 2000 platform (Illumina). Analysis and filtering were done as described previously [15].
For family 3, WES was performed on the two affected siblings (patients 3 and 4) and the mother at Ambry Genetics (Aliso Viejo, CA). Exome library preparation, sequencing, bioinformatics, and data analyses were performed as previously described [16]. Briefly, samples were prepared and sequenced using paired-end, 100 cycle chemistry on the Illumina HiSeq 2000. Exome enrichment was performed using the Agilent SureSelect Target Enrichment System (Agilent Technologies). Data were annotated with the Ambry Variant Analyzer tool.
For family 4, WES was performed on genomic DNA extracted from leukocytes of patient 5 and both parents. Exome enrichment was performed using the Nextera Enrichment Kit (37 Mb) (Illumina) and sequencing on the HiSeq4000 platform (Illumina). Variant analysis was done as previously described [17].
For family 5, WES was performed on leukocyte-derived genomic DNA in patient 7 using Illumina’s Nextera Rapid Capture Exome Kit on the Illumina NextSeq Platform (Illumina). The average coverage depth was 130×, with ~95% of the bases covered at >20×, and a sensitivity of >90% [18]. Data were stored and analyzed using a previously published automated pipeline, SeqMule v1.2.5 [19]. The variant call format file was annotated by ANNOVAR v.2016Feb01 [20]. Variants were filtered to 1% allele frequency in population databases including ExAC, 1000 genome database, and an internal database of exome sequences of 405 individuals of Indian origin. Exonic and splice site variants were then prioritized by MIM (Mendelian Inheritance in Man) identity, phenotypic assessment, and the American College of Medical Genetics (ACMG) criteria of pathogenicity [21].
The variants identified were described according to HGVS nomenclature [22], using the reference sequence NM_004037.7 and were submitted to the Leiden Open Variation Database (https://databases.lovd.nl/shared/genes/AMPD2) (patient IDs 00133645, 00133661-00133664).
Variant validation
AMPD2 (NM_004037.7) variant validation and segregation analysis in patients 1–5 and 7, their parents (or mother of patient 8) and healthy siblings were performed by Sanger-sequencing. Exons 6 and 19 harboring the AMPD2 variants detected in patient 5 were analyzed in patient 6, the similarly affected brother, by Sanger-sequencing. The coding exons and surrounding intronic sequences of the AMPD2 gene were PCR-amplified from leukocyte-derived DNA of patient 8 and sequenced. Amplicons were directly sequenced using the ABI BigDye Terminator Sequencing kit (Applied Biosystems) and an automated capillary sequencer (ABI 3500, Applied Biosystems). Sequence electropherograms were analyzed using the Sequence Pilot software (JSI Medical Systems).
Results
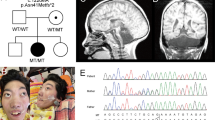
We performed WES in six individuals from five unrelated families with severe global developmental delay in all (patients 1–5 and 7) and postnatal microcephaly in patients 1, 2, 4, 5 and 7 (Table 1). Sanger-sequencing of AMPD2 was performed in patient 8 due to microcephaly and PCH9-typical brain malformations identified by MRI (see below). Parents of the unrelated patients 1, 2, 7 and 8 were consanguineous (Table 1). Brain imaging in six of the seven patients revealed pontocerebellar hypoplasia of variable severity, severe to mild hypoplasia of the corpus callosum and atrophy of the cerebral cortex as consistent findings (Table 1 and Fig. 1). We identified rare biallelic variants in the AMPD2 gene in all affected individuals: in patient 1, we found the homozygous 13-bp duplication c.1424_1436dup, predicting a frameshift and introduction of a premature termination codon (p.(Asp480Glyfs*13)), in patient 2 the homozygous missense variant c.1648G>A [p.(Glu550Lys)], in the two affected siblings (patients 3 and 4) the two heterozygous missense variants c.1133G>C/p.(Arg378Pro) and c.1232A>G/p.(Asn411Ser), in patient 5 the heterozygous nonsense variant c.682G>T/p.(Glu228*) and the heterozygous missense variant c.2528G>A/p.(Arg843His), and in patients 7 and 8 the homozygous missense variant c.1132C>T/p.(Arg378Trp) and c.2128G>A/p.(Gly710Arg), respectively (Table 1, Fig. 2 and Supplementary Figure 1). Segregation analysis in the families confirmed each of the parents as heterozygous carrier of an AMPD2 variant, except of patient 8’s parents as only the mother was available (Fig. 2 and Supplementary Figure 1). The 3-month-old brother of patient 5 showed deceleration of head growth (patient 6 in Table 1), and fetal MRI already had revealed pontocerebellar hypoplasia, a nearly absent corpus callosum, and enlarged ventricles (Fig. 3). Testing of the two familiar AMPD2 variants c.682G>T and c.2528G>A in patient 6 revealed both sequence alterations (Table 1, Fig. 2 and Supplementary Figure 1). The three variants c.1424_1436dup, c.1648G>A and c.1232A>G were absent from the gnomAD browser (~250,000 alleles); the c.1132C>T change has a minor allele frequency of 0.00001219, the c.2128G>A transition of 0.000008230, the c.1133G>C change of 0.000004063, the c.682G>T variant of 0.00002599, and the c.2528G>A change of 0.000004066 in the gnomAD browser, but none was listed in the homozygous state (Supplementary Table 1). All AMPD2 alterations were computationally predicted to be deleterious, and classification of the variants according to the ACMG guidelines [21] revealed the AMPD2 frameshift, the nonsense and the p.(Arg843His) change to be “pathogenic” and p.(Arg378Pro), p.(Arg378Trp), p.(Asn411Ser), p.(Glu550Lys), and p.(Gly710Arg) as “likely pathogenic” variants (Supplementary Table 1).

Magnetic resonance imaging characteristics of patients 1–3, 5, 7, and 8. a–d Patient 1: a Sagittal cut through the midline showing almost complete absence of the corpus callosum, with absent cingulate gyrus and pontocerebellar hypoplasia, involving the cerebellar vermis and hemispheres (asterisk). b Coronal cut showing dilated subarachnoid spaces and lateral ventricles with a typical moose head sign. c Axial cut through the midbrain showing its typical “figure 8” appearance (encircled). d Axial cut through the lateral ventricles showing their dilated shape, with a wide communication with the third ventricle, and protruding thalami. T2-weighted images a, b, d show hypomyelination and enlarged cortical sulci. e–h Patient 2: e Sagittal cut through the midline showing almost complete absence of the corpus callosum, with a dysmorphic cingulate gyrus, and pontocerebellar hypoplasia (asterisk). f Coronal section showing dilated ventricles and cerebellar hypoplasia. g Axial section through the midbrain showing a hypoplastic “figure 8” shape (encircled). h Axial cut through the lateral ventricles showing their dilated shape, with posterior colpocephalic aspect, the wide communication with the third ventricle, and protruding thalami. T2-weighted images e, g, h document hypomyelination and dilated cortical sulci. i–l Patient 3: i Sagittal cut through the midline showing a thin corpus callosum underlying a regular cingulate gyrus, and mild cerebellar vermis hypoplasia (asterisk). j Coronal section well demonstrating the atrophic cerebral cortex with dilated subarachnoid spaces. k, l Axial sections showing mild midbrain hypoplasia k, which is not severe enough to develop a “figure 8” shape, and an almost normal shape and size of the lateral ventricles l. Hypomyelination is apparent in the T2-weighted images j, k. m–p Patient 5: m Sagittal cut showing an extremely thin corpus callosum, overlaid by an underdeveloped cingulate gyrus, and pontocerebellar hypoplasia (asterisk). n Coronal section showing dilated ventricles and cerebellar hypoplasia. o Axial section through the midbrain showing its hypoplastic “figure 8” shape (encircled). p Axial cut through the lateral ventricles showing their dilated shape, with posterior colpocephaly, a wide communication with the third ventricle and protruding thalami. Hypomyelination is apparent in T2-weighted images m, p. q–t Patient 7: q Sagittal cut showing an extremely thin corpus callosum, overlaid by a cingulate gyrus with radially oriented sulci, and pontocerebellar hypoplasia (asterisk). r Coronal section showing dilated ventricles, cortical atrophy and cerebellar hypoplasia. s Axial section through the midbrain showing its hypoplastic “figure 8” shape (encircled). t Axial section showing dilated cortical sulci and posterior horns of the lateral ventricles. u–x Patient 8: u Sagittal cut showing mild pontocerebellar hypoplasia (asterisk) and an extremely thin corpus callosum, whose size and shape are comparable with those observed in patients 1, 2, 5, and 7. v Coronal section showing mild cortical and cerebellar atrophy. w Axial section through the midbrain showing a “figure 8” shape (encircled). x Axial section showing dilated cortical sulci and protruding thalami

Pedigrees of the six studied families and segregation analysis of the AMPD2 variants in the eight patients (P1 in a; P2 in b; P3 and P4 in c; P5 and P6 in d; P7 in e; P8 in f) and their healthy parents (or healthy mother of P8). Black symbols indicate individuals with PCH9. NA: not available for testing

Fetal MRI of patient 6 at 24 weeks’ gestation. These images show pontocerebellar hypoplasia (sagittal in a) and an extremely thin, nearly absent corpus callosum (a and coronal in b), with dilated ventricles with colpocephalic presentation (axial in c). The coronal cut b reveals the typical moose head appearance of the lateral and third ventricles. Note for comparison the morphologic differences with images taken at approximately the same levels in a normal fetus (control) of the same gestational age d–f
Occipital frontal circumference (OFC) at birth was normal or low normal in patients 1, 5, 6, and 8. Seven of the eight patients developed severe postnatal microcephaly (−4.1 to −6.8 SD between the age of 11 months and 6 years and 3 months). Patient 3 did not have microcephaly at the age of 4 years and 2 months (Table 1). Earlier measurements of her head circumference also were in the low-normal range with 47.5 cm (−1.3 SD) at 2 years 6 months and 48 cm (−1.5 SD) at 3 years 6 months.
Detailed clinical data were available for patients 1–5, 7, and 8 (Table 1). At the age of 2 months, patient 1 exhibited respiratory insufficiency with severe apnea that persisted during the first year of life; he suddenly died at the age of 13 months, likely due to prolonged apnea during sleep. Facial dysmorphism was noted in patients 1, 3–5, and 7 (Table 1). Only one of seven patients had seizures, but all exhibited irritability (see also EEG data in Table 1). Early hypotonia was present in six patients, five later showed hypertonia. Lack of visual contact (7/7), central visual impairment (5/6), pale optic disc (2/7) and primary optic atrophy (1/7) were observed in our patient cohort. An abnormal posture with opisthotonus and flexion of the arms were additional findings in three patients with hypertonia. Spasticity in patients 7 and 8, dyskinesia in patient 2, chorea and “handwashing” movements in patients 3 and 4, and dystonic movements of the head in patient 8 were also reported. There was no development at all in patients 1, 7, and 8, while motor and cognitive development was delayed or severely delayed in the remaining four individuals. Metabolic findings in patient 3 were mitochondrial complex I deficiency and abnormal oxidative phosphorylation enzymology on mitochondria isolated from fresh muscle. These findings were unexpected as no mitochondrial respiratory chain defects have yet been reported in patients with PCH9 [6,7,8,9]. There are two possible explanations: either patient 3 has complex I deficiency or the results of this assay are false positive. WES did not reveal any variant affecting function in nuclear genes implicated in mitochondrial complex I (data not shown). However, we cannot exclude some type of secondary mitochondrial dysfunction. Other clinical manifestations include sleep disturbance, postprandial hyperglycemia and gastroesophageal reflux in patient 1. At latest examination, the 6-year-old patient 2 was wheelchair-bound and showed severe developmental delay, but could communicate nonverbally, started to fixate and had social contact; he exhibited moderate hearing impairment, a thyroglossal cyst and was fed by G-tube. Hypophyseal dysfunction and clubfeet were present in patient 5 (Table 1).
Brain MRI was performed in seven of the eight patients (MRI scans of patient 4 are not available) and showed overlapping structural abnormalities in all, with variable severity (Figs. 1 and 3). Neuroimaging was performed three times in patient 1, at the age of 16 days, 3 months and 8 months. The first MRI scan revealed partial agenesis of the corpus callosum and hypoplasia of the pons, brainstem, cerebellar vermis and cerebellar hemispheres. While these anomalies were not found to be progressive, cortical atrophy, ventricular dilatation, and hypomyelination became prominent, especially in the latest MRI scans (Fig. 1a–d). Patient 2 had a first MRI scan at the age of 3 months that revealed severe callosal hypoplasia, pontocerebellar hypoplasia, and delayed myelination. A follow-up brain imaging at the age of 11 months demonstrated increased CSF spaces supratentorially and infratentorially and severe myelination delay; however, there was no progression of cerebellar and brainstem anomalies (Fig. 1e–h). Patient 3 was imaged only once, at the age of 1 year. A thin hypoplastic corpus callosum, especially in its posterior part, mild pontocerebellar hypoplasia and generalized atrophy of the cerebral cortex were identified; hypomyelination was also present (Fig. 1i–l). Brain imaging in patient 5 was performed at age 5 months and repeated at 3 years. Patient 7 had brain imaging at age 4 years and patient 8 at 6 months. MRIs of the three individuals exhibited structural changes that were similar to those observed in patients 1 and 2, including an extremely thin corpus callosum, pontocerebellar hypoplasia, a small midbrain and dilated lateral ventricles with mildly progressive cortical atrophy. T2-weighted images also demonstrated hypomyelination in patients 5 and 7; the early MRI scan in patient 8 could not clearly detect hypomyelination (Fig. 1m–x). In patient 6, fetal MRI, performed at 24 weeks of gestation, disclosed early morphological changes consistent with those characterizing postnatal imaging in the other five patients, including an extremely thin corpus callosum, pontocerebellar hypoplasia, and dilated ventricles with a colpocephalic configuration (Fig. 3). The unique finding in MRI scans, a “figure 8” appearance of the brainstem on axial images [6], was demonstrated in patients 1, 2, 5, 7, and 8, but was absent in patient 3 (Fig. 1c, g, k, o, s, w).
Discussion
AMPD2 encodes one of three paralogous AMP deaminases (AMPD1, AMPD2, and AMPD3) catalyzing the irreversible, hydrolytic deamination of AMP to IMP and NH3. The C-terminal domains that contain the catalytic domain and an ATP binding site are conserved between the paralogs, whereas the N-terminal regions are highly divergent [23]. The nine previously published and the seven novel AMPD2 variants likely affecting function reported here, all found in individuals with autosomal recessively inherited PCH9, comprise three nonsense, three frameshift, and ten missense variants affecting highly conserved amino acid residues. Nine of the 10 missense variants are located in exons 8–17 encoding the AMP deaminase domain (Fig. 4) and likely represent loss-of-function alleles [6, 7, 24]. However, the p.(Arg251Trp) change is located outside the catalytic domain (Fig. 4), but destabilizes the AMPD2 protein leading to its degradation [8]. AMPD deficiency in patient-derived neural progenitor cells has been shown to cause vulnerability to physiological levels of adenosine resulting in a significantly reduced cell viability and thereby recapitulating the neurodegenerative phenotype in PCH-affected individuals [6].

Schematic representation of the exon-intron structure of six alternative AMPD2 transcripts (exons are numbered according to Fig. 1 in reference 28 and Fig. 3 in reference 7). Boxes represent exons and lines introns. Coding (parts of) exons are indicated in dark gray and untranslated regions in light gray. The respective mRNA reference sequence number of each transcript is given above or in front of the transcripts. PCH9-associated variants identified in this study are indicated in red, previously reported variants in dark blue. The predicted consequence of variant c.495delG on protein level has been corrected from p.(Arg165fs*21) [9] to p.(Gln166Argfs*21). The single frameshift variant found in a family with spastic paraplegia type 63 is shown in light blue; this 1-bp deletion affects the 5′ untranslated region of one and the coding region of three out of six transcripts. A yellow box frames the exons encoding the adenosine monophosphate (AMP) deaminase domain
A total of 25 individuals with biallelic AMPD2 variants and a severe neurodevelopmental phenotype have been clinically studied to date (Patients 1–8 in this report and 17 published PCH9-affected individuals in Table 2 [6,7,8,9]; the number of patients in whom a specific clinical feature is present is given as the proportion of those patients where information was available). Vanderver et al. (2016) reported the homozygous AMPD2 variant p.(Arg843His) in a 4-year-old girl (LD_0673) [24]. In spite of the diagnosis of spastic paraplegia she presented with microcephaly, myoclonic epilepsy, severe hypotonia, global developmental delay and volume loss of the brainstem and cerebellar hemispheres on brain imaging [24] strongly suggesting PCH9. In addition, the amino acid change p.(Arg843His) has also been found in patients 5 and 6 reported here (Table 1). In 18 patients, OFC at birth was within the normal to low-normal range (Table 2). Twenty four of 25 patients (96%) developed severe microcephaly (−3 to −9 SD), and cognitive development was profoundly or severely delayed in 24/24. An exception is patient 3 reported here as her cognitive and motor development was delayed, but not profoundly or severely, she did not show microcephaly, and her brain malformations were mild compared with other patients (Table 1). These findings suggest that protective genetic variants [25] and environmental modifiers can exert their effects on the phenotype of individuals with biallelic AMPD2 variants.
Different seizure types were observed in 58% (14/24) of the patients (Table 2). They frequently exhibited spasticity, hypotonia and hypertonia (92, 72, and 86%, respectively). 83% (19/23) of the patients had central visual impairment and 53% (8/15) primary optic atrophy or pale optic disc. Electrophysiologic studies revealed peripheral nervous system manifestations in two of the four patients tested (Table 2). However, as axonal neuropathy is developing with age, this feature may be absent in infants and young children with biallelic AMPD2 variants. Common dysmorphic features including large and posteriorly rotated ears, mandibular hypoplasia and mottled and fragile teeth with multiple cavities were noted in three affected siblings [9]. Although craniofacial dysmorphism has also been reported in some other patients with PCH9, the features, except microcephaly with sloping forehead, were not consistent between unrelated individuals or even absent (see also patients 1–5, 7, and 8 in Table 1) [7, 9].
MRI scans showed pontocerebellar hypoplasia (24/24), generalized atrophy of the cerebral cortex (10/10), hypoplasia or aplasia of the corpus callosum (24/24) and hypomyelination (11/15) as the most consisting findings (Table 2). The typical brain imaging finding was the “figure 8” appearance of the midbrain (17/18); however, this characteristic midbrain shape was absent on the axial MRI of patient 3 reported here (Fig. 1). The proportionate involvement of cerebellar vermis and hemispheres in patients with AMPD2 variants may help distinguishing them from PCH2-affected or PCH4-affected individuals, who show flat cerebellar hemispheres and a less severely affected vermis (dragonfly-like appearance of the cerebellum on coronal images) [2, 26]. Although corpus callosum hypoplasia has also been reported in PCH type 8 [27], the combination of callosal hypoplasia or aplasia and “figure 8” appearance of the midbrain should prompt AMPD2 testing in infants and children with developmental delay and PCH with or without microcephaly (as demonstrated for patient 8 in our cohort). However, mild hypoplasia of cerebellum, pons, brainstem, and corpus callosum and absence of the “figure 8” midbrain shape in patient 3 reported here (Fig. 1) indicate that MRI clues which help distinguishing between different PCH types can be absent.
Novarino et al. [11] reported the homozygous 1-bp deletion c.319delT/p.(Cys107Alafs*80) in AMPD2 (previously reported as c.318delT/p.C107Afs365X) in a 20-year-old female and her 5-year-old nephew with a complicated form of hereditary spastic paraplegia (HSP). Both walked unsupported, had normal cognition, and MRI of the male individual did not show cerebellar or brainstem abnormalities [11]. These findings raised the question of how variants in AMPD2 can cause both a severe PCH disorder and a relatively mild form of HSP. The c.319delT variant affects the coding region of three of the six alternative AMPD2 transcripts generating five different AMP deaminase isoforms. The AMPD2 transcript variants that differ at their 5’ ends are produced through the regulation of five separate promoters and a cassette-type alternative splicing event involving exon 2 (Fig. 4) [23]. Five alternative AMPD2 mRNAs have been studied in detail (Fig. 4): The AMPD2 transcript with the exon 1A-2 configuration (NM_139156.3) is predominantly expressed in liver, while the AMPD2 mRNAs with the exon 1B-2 (NM_004037.7, NM_001257360.1), exon 1B-3 (NM_001308170.1) and exon 3–4 configuration (lacking three alternate 5’ exons; NM_203404.1) are mainly expressed in brain [23,28,29,30]. As the c.319delT mutation is located in the alternatively spliced exon 2, only two of the four AMPD2 transcript variants expressed in brain (NM_004037.7 and NM_001257360.1) are affected. Accordingly, high expression of the two alternative AMPD2 mRNAs without exon 2 in brain (NM_001308170.1 and NM_203404.1) might have compensated for loss-of-function of the exon 2-containing mRNAs that possibly caused motor neuron degeneration in the two relatives but left the brain intact. In contrast, all AMPD2 variants associated with the severe PCH9 phenotype are located in exons containing coding information for all AMPD isoforms and affect each of the six transcript variants (Fig. 4). Consistent with this, Marsh et al. [8] have recently discussed functional redundancy of AMPD2 isoforms and AMPD paralogs to underlie the different human disease presentations. A remarkably similar molecular mechanism could be proposed for biallelic variants in the EXOSC3 gene that cause both PCH1 and a complicated form of HSP without pontine hypoplasia [31,32,33]. The homozygous missense variant c.571 G >T/p.(Gly191Cys) reported in a family with four HSP-affected members [32] is located in the alternatively spliced exon 3 of EXOSC3 (NM_016042.3), while the majority of the PCH1-associated variants is present in exon 1 or 2 and affect the two alternative EXOSC3 mRNAs (NM_016042.3 and NM_001002269.2). Altogether, these data may provide preliminary evidence that functional redundancy of one or several AMPD2 or EXOSC3 isoforms in the brain explains the neurodegenerative disease affecting only motor neurons, while deficiency of all isoforms has a combined effect on hindbrain and motor neurons. In case of AMPD2, the divergent N-terminal regions which play a critical role for the different isoforms to function under different metabolic conditions in various tissues and organs [10, 30, 34] may help explaining the observed variability in disease expression. Similarly, Ampd3 compensates for loss of Ampd2 in mice as Ampd2-deficient mice show proteinuria, a nephrotic syndrome and hypercholesterolemia [35, 36], whereas Ampd2 and Ampd3 double-knockout mice model the PCH9 phenotype [6]. We are not aware of any renal problems in our and previously reported patients with AMPD2 variants.
Most PCH types exhibit variants in genes that encode proteins involved in RNA metabolism, such as tRNA processing (TSEN2, TSEN15, TSEN34, TSEN54, and CLP1) [26,37,38,39], tRNA synthesis (RARS2) [40] and RNA maturation and surveillance (EXOSC3) [41, 42], nucleotide metabolism, for example GTP synthesis (AMPD2) [6], and amino acid biogenesis (SEPSECS) [43]. The disease gene for PCH type 7, TOE1, underscores the importance of RNA processing for midbrain and hindbrain maturation as the encoded deadenylase is important for small nuclear RNA processing [44]. In contrast, TBC1D23, a recently reported disease gene for a non-degenerative PCH form (PCH type 11; MIM 617695), encodes a RAB-specific GTPase-activating protein implicated in intracellular vesicle trafficking [4, 5]. A protein synthesis defect identified in AMPD2-deficient cells and a yeast strain with a mutation in sen2, the ortholog to human TSEN2, suggests that impaired protein translation may be a general mechanism underlying the neurodegenerative phenotype in many PCH types [6]. This assumption is further supported by the finding of progressive neurodegeneration in mice with loss of a protein that functions in the release of stalled ribosomes during translation (GTPBP2). However, deficiency of GTPBP2 only caused the neurodegenerative phenotype in a mouse strain bearing a mutation in the brain-specific tRNA for arginine [45]. Taken together, excessive ribosome stalling induced by loss of two factors important for efficient protein translation reveals a link between defects in the protein synthesis machinery and neuronal degeneration [46, 47]. However, the work of Ishimura et al. (2014) also demonstrates the importance of the genetic background on phenotypic expression, not only in mice but also in humans [45]. Accordingly, the wide spectrum of neurological phenotypes associated with variants in the classical PCH genes may not only be explained by functional redundancy of brain-specific isoforms but also by the impact of yet to be identified modifying genes.
References
Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–22.
Namavar Y, Barth PG, Kasher PR, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134:143–56.
Rudnik-Schöneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C. 2014;166C:173–83.
Ivanova EL, Mau-Them FT, Riazuddin S, et al. Homozygous truncating variants in TBC1D23 cause pontocerebellar hypoplasia and alter cortical development. Am J Hum Genet. 2017;101:428–40.
Marin-Valencia I, Gerondopoulos A, Zaki MS, et al. Homozygous mutations in TBC1D23 lead to a non-degenerative form of pontocerebellar hypoplasia. Am J Hum Genet. 2017;101:441–50.
Akizu N, Cantagrel V, Schroth J, et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell. 2013;154:505–17.
Marsh AP, Lukic V, Pope K, et al. Complete callosal agenesis, pontocerebellar hypoplasia, and axonal neuropathy due to AMPD2 loss. Neurol Genet. 2015;1:e16.
Marsh AP, Yap P, Tan T, et al. A novel AMPD2 mutation outside the AMP deaminase domain causes pontocerebellar hypoplasia type 9. Am J Med Genet A. 2017;173:820–823.
Accogli A, Iacomino M, Pinto F, et al. Novel AMPD2 mutation in pontocerebellar hypoplasia, dysmorphisms, and teeth abnormalities. Neurol Genet. 2017;3:e179.
Bausch-Jurken MT, Sabina RL. Divergent N-terminal regions in AMP deaminase and isoform-specific catalytic properties of the enzyme. Arch Biochem Biophys. 1995;321:372–380.
Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511.
Girisha KM, Kortüm F, Shah H, et al. A novel multiple joint dislocation syndrome associated with a homozygous nonsense variant in the EXOC6B gene. Eur J Hum Genet. 2016;24:1206–1210.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Spielmann M, Kakar N, Tayebi N, et al. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res. 2016;26:183–191.
Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–586.
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–182.
Girisha KM, Shukla A, Trujillano D, et al. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016;90:536–539.
Guo Y, Ding X, Shen Y, Lyon GJ, Wang K. SeqMule: automated pipeline for analysis of human exome/genome sequencing data. Sci Rep. 2015;5:14283.
Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158.
Richards S, Aziz N, Bale S, et al. Standards and guideline`s for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37:564–569.
Sabina RL, Mahnke-Zizelman DK. Towards an understanding of the functional significance of N-terminal domain divergence in human AMP deaminase isoforms. Pharmacol Ther. 2000;87:279–283.
Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79:1031–1037.
Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531–538.
Budde BS, Namavar Y, Barth PG, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113–1118.
Mochida GH, Ganesh VS, de Michelena MI, et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat Genet. 2012;44:1260–1264.
Bausch-Jurken MT, Mahnke-Zizelman DK, Morisaki T, Sabina RL. Molecular cloning of AMP deaminase isoform L. Sequence and bacterial expression of human AMPD2 cDNA. J Biol Chem. 1992;267:22407–22413.
Mahnke-Zizelman DK, van den Bergh F, Bausch-Jurken MT, et al. Cloning, sequence and characterization of the human AMPD2 gene: evidence for transcriptional regulation by two closely spaced promoters. Biochim Biophys Acta. 1996;1308:122–132.
Van den Bergh F, Sabina RL. Characterization of human AMP deaminase 2 (AMPD2) gene expression reveals alternative transcripts encoding variable N-terminal extensions of isoform L. Biochem J. 1995;312(Pt 2):40410.
Eggens VR, Barth PG, Niermeijer JM, et al. EXOSC3 mutations in pontocerebellar hypoplasia type 1: novel mutations and genotype-phenotype correlations. Orphanet J Rare Dis. 2014;9:23.
Halevy A, Lerer I, Cohen R, et al. Novel EXOSC3 mutation causes complicated hereditary spastic paraplegia. J Neurol. 2014;261:2162169.
Rudnik-Schöneborn S, Senderek J, Jen JC, et al. Pontocerebellar hypoplasia type 1: clinical spectrum and relevance of EXOSC3 mutations. Neurology. 2013;80:438–446.
Haas AL, Sabina RL. N-terminal extensions of the human AMPD2 polypeptide influence ATP regulation of isoform L. Biochem Biophys Res Commun. 2003;305:421–427.
Helmering J, Juan T, Li CM, et al. A mutation in Ampd2 is associated with nephrotic syndrome and hypercholesterolemia in mice. Lipids Health Dis. 2014;13:167.
Toyama K, Morisaki H, Cheng J, et al. Proteinuria in AMPD2-deficient mice. Genes Cells. 2012;17:28–38.
Breuss MW, Sultan T, James KN, et al. Autosomal-recessive mutations in the tRNA splicing endonuclease subunit TSEN15 cause pontocerebellar hypoplasia and progressive microcephaly. Am J Hum Genet. 2016;99:228–235.
Karaca E, Weitzer S, Pehlivan D, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650.
Schaffer AE, Eggens VR, Caglayan AO, et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157:651–663.
Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–862.
Kilchert C, Wittmann S, Vasiljeva L. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol. 2016;17:227–239.
Wan J, Yourshaw M, Mamsa H, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708.
Agamy O, Ben Zeev B, Lev D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. 2010;87:538–544.
Lardelli RM, Schaffer AE, Eggens VR, et al. Biallelic mutations in the 3’ exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing. Nat Genet. 2017;49:457–464.
Ishimura R, Nagy G, Dotu I, et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459.
Darnell JC. Molecular biology. Ribosome rescue and neurodegeneration. Science. 2014;345:378–379.
Kapur M, Monaghan CE, Ackerman SL. Regulation of mRNA translation in neurons-A matter of life and death. Neuron. 2017;96:616–637.
Acknowledgements
We are grateful to the families who contributed to this study. We would like to thank Inka Jantke for skillful technical assistance and Jelena Bircic and Anna Podolska for help with WES data interpretation and segregation analysis in family 1. This work was supported by grants from the Deutsche Forschungsgemeinschaft (KO 4576/1–2 to F.K. and KU 1240/10–1 to K.K.), the Department of Health Research [project “Clinical and molecular characterization of leukodystrophies in Indian children” (V.25011/379/2015-GIA/HR)], and the EU 7th Framework Programme (FP7) under the project DESIRE (N602531 to R.G.).
Author contributions
F.K. performed WES, WES data analysis and interpretation of sequence variants in patient 1, segregation analysis by Sanger-sequencing, and drafting of Figs. 2 and 4, Table 1, supplementary figure 1 and supplementary table 1; R.A.J. performed WES data analysis and interpretation of sequence variants in patients 5 and 6, segregation analysis by Sanger-sequencing, and wrote the manuscript; M.A. performed WES data processing for patient 1; G.B. performed WES data analysis and interpretation of sequence variants in patient 2, segregation analysis by Sanger-sequencing, and wrote the manuscript; K.L.H. and S.T. performed WES data analysis and interpretation of sequence variants in patients 3 and 4, and wrote the manuscript; M.H. performed WES data analysis and interpretation of sequence variants in patient 7, segregation analysis by Sanger-sequencing, and wrote the manuscript. S.A.B., D.H., G.C.K., A.S., K.M.G., M.S., and S.W.-W. conducted clinical phenotyping; M.M. and R.G. evaluated all MRI scans, drafted Figs. 1 and 3, and wrote the manuscript; J.R.L. and K.K. initiated and directed the project, drafted Table 2, and wrote the manuscript; all authors discussed the results and commented on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
K.L.H. was and S.T. is employed by and receive a salary from Ambry Genetics; whole-exome sequencing is among its commercially available tests. D.H. is employed by and receives a salary from MVZ Labor Leipzig. R.G. has received travel funding and honoraria for Advisory Board activities from Eisai, Inc., Novartis, and Zogenix; has received travel funding from UCB; has served on the editorial boards of Epilepsia, Progress in Epileptic Disorders, Neuropediatrics, the Journal of Child Neurology, Seizure, BMC Medical Genetics, Topics in Epilepsy, the Journal of Pediatric Epilepsy, Epileptic Disorders, the European Neurological Journal, Neurology, and the Journal of Embryology and Developmental Biology; receives publishing royalties from Cambridge University Press, Lippincott Williams and Wilkins, John Libbey Eurotext, and Oxford University Press; and has received research support from the European Union, Tuscany Region Research Department, EC, Italian Ministry of Health and, and the Pisa Foundation. The other authors report no disclosures relevant to the manuscript.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kortüm, F., Jamra, R.A., Alawi, M. et al. Clinical and genetic spectrum of AMPD2-related pontocerebellar hypoplasia type 9. Eur J Hum Genet 26, 695–708 (2018). https://doi.org/10.1038/s41431-018-0098-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0098-2
This article is cited by
-
Demographic and clinical characteristics, seizure disorders, and antiepileptic drug usage in different types of corpus callosum disorders: a comparative study in children
Italian Journal of Pediatrics (2024)
-
Broadening the phenotype and genotype spectrum of novel mutations in pontocerebellar hypoplasia with a comprehensive molecular literature review
BMC Medical Genomics (2024)
-
Role of AMP deaminase in diabetic cardiomyopathy
Molecular and Cellular Biochemistry (2024)
-
Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study
The Cerebellum (2024)
-
Pontocerebellar hypoplasia type-9 due to a novel p.Arg503Ter truncating variant in AMPD2: a report from India
Acta Neurologica Belgica (2023)
