
Abstract
Kinesin proteins are critical for various cellular functions such as intracellular transport and cell division, and many members of the family have been linked to monogenic disorders and cancer. We report eight individuals with intellectual disability and microcephaly from four unrelated families with parental consanguinity. In the affected individuals of each family, homozygosity for likely pathogenic variants in KIF14 were detected; two loss-of-function (p.Asn83Ilefs*3 and p.Ser1478fs), and two missense substitutions (p.Ser841Phe and p.Gly459Arg). KIF14 is a mitotic motor protein that is required for spindle localization of the mitotic citron rho-interacting kinase, CIT, also mutated in microcephaly. Our results demonstrate the involvement of KIF14 in development and reveal a wide phenotypic variability ranging from fetal lethality to moderate developmental delay and microcephaly.
Similar content being viewed by others

Introduction
KIF14 (Kinesin family 14, MIM: 611279), encodes a member of the kinesin-3 superfamily of microtubule motor proteins which act predominantly as “plus-end” directed molecular motors that generate force and movement across microtubules [1, 2]. Kinesin proteins were first identified in 1985 [3, 4] and their action is critical for numerous cellular functions such as intracellular transport and cell division [5, 6]. Fourteen members of the kinesin family have so far been linked with various degrees of evidence to monogenic disorders with several modes of inheritance, and a wide phenotypic spectrum affecting several organ systems (Table 1). KIF14 has a major role in cytokinesis and has been extensively studied in cancer [7]. Somatic variants have classified KIF14 as an oncogene [8]; it has been suggested that its overexpression could be used as a prognostic marker in various tumor types [9, 10] and is a potential target for cancer treatment [11, 12].
Numerous studies have relied on the power of consanguineous families to identify causes of recessive disease by applying homozygosity mapping strategies, or by filtering genome-wide data sets for the restricted set of homozygous deleterious variants. The accessibility of exome and genome variomes capturing extant genetic variation in special populations, mostly from Asia and the Middle East, has allowed prioritization of variants that are rare at the population level, and thus more likely to contribute to disease [13].
As part of an effort to identify new recessive syndromes in consanguineous pedigrees, we present here four families with intellectual disability/developmental delay (ID/DD), small head size, and a variety of additional clinical symptoms. Interestingly, a mouse model, laggard (lag), carrying a spontaneous variant [14] has been shown to have a similar phenotype with growth retardation, brain size reduction, severe hypomyelination of the central nervous system including the optic nerve, and motor impairment. Lag/lag mice die before weaning and targeted knockouts have confirmed KIF14 as the causative gene. In addition, a single family was described by Filges et al. [15]. with two fetuses showing identical severe intrauterine growth restriction (IUGR), microcephaly, brain malformations, renal cystic dysplasia/agenesis, and other genitourinary malformations, that displayed biallelic deleterious variants in KIF14. An autosomal recessive disease with lethal fetal ciliopathy phenotype was proposed.
Materials and methods
Patient description
Family 1
Family 1 (Fig. 1) is of Egyptian origin and the patients were offspring of first cousins, the father being himself an offspring of a consanguineous marriage. The older affected girl was born after a non-remarkable 40-week pregnancy. Birth measurements are reported as average. At her first clinical exam at 12 years of age, she weighted 18 kg (−2.6 SD), the head circumference was 52 cm (−1.3 SD), and height was 125 cm (−4 SD). She sat at 4 years, has never walked and while at birth she was described as hypotonic, at the time of her clinical examination her muscle tone was spastic with brisk reflexes. She has never spoken and is suffering from severe ID. G-band karyotype (46,XX) was normal as well as the rest of her work-up: brain CT, metabolic screening, thyroid function tests, hearing test, and complete eye evaluation.

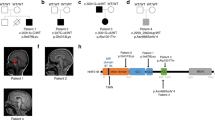
Family pedigrees, genotype, and KIF14 variants. a Pedigrees of four families affected by KIF1 variants showing segregation of the altered alleles and chromatograms of these variants-altered alleles shown by “+” and wild-type allele shown by “−”. b, c Schematic overview of the KIF14 gene (top) and protein (bottom) and location of the identified variants in the current study(black) and two compound variants from previous study (gray)
The younger affected sister was also born after an uneventful full-term pregnancy, birth measurements are not available. At 3 years of age she weighed 10.5 kg (−2 SD), had a height of 82 cm (−2.9 SD) and a head circumference of 47 cm (−1.6 SD). According to the parents, she has an identical clinical course to that of her older sister with ID/DD. Brain CT, thyroid/metabolic screening, and hearing/vision gave unremarkable results, like her sister. Both sisters had small mouth, wide and sparse eyebrows, and strabismus. The older sister also has a characteristic foot morphology with big first toe, sandal gap, external deviation of the 2nd and 3rd toes, the latter being overlapped by the 4th toe. The younger sister has no feet anomalies.
Family 2
Family 2 is of Turkish origin, and the parents are first cousins. Two out of five siblings are affected with severe ID, microcephaly, complete blindness with optic nerve hypoplasia, and micropthalmia. The proband in this family was born in Turkey after an uneventful pregnancy and had a normal birth weight. The parents discovered his blindness at 3 weeks of age. He was evaluated the Pediatric Department in Oslo at 7 years of age when the family arrived in Norway; he was blind with bilateral microphthalmia, had severe ID/DD and no language, severe microcephaly, could stand and walk with support. He had been exclusively milk-fed and was cachectic. Head CT was normal at 7 years of age, and MRI at 8 years of age showed reduced brain volume, large basal cisterns, and optic atrophy. A pathological EEG pattern during sleep was documented, with frequent sharp focal activity, and occasional spike and wave, left temporal-occipital region, but no seizures. He had normal reflexes on neurological exam. At 14 years of age, his OFC was 48 cm, length 145 cm (−3.4 SD), and weight was only 28 kg. At 23 years of age, his OFC was still the same (−5.1 SD), his length 157 cm (−3.7 SD), and weight 47 kg. He has bilateral microphthalmos and extreme microcephaly, otherwise normal facial features. He has severe behavioral problems with aggressive outbursts and violence against caregivers, that are often triggered by certain sounds or noise, and he has self-stimulating and self-destructive behavior. He is autistic, has tactile aversion and is sound sensitive, but enjoys classical music and is surprisingly well oriented in the examination room.
His younger, affected sister (Fig. 1, individual IV:6) was born at term with Apgar 9/9, birth weight 3450 g, birth length 49 cm, and had OFC 32 cm (−2.6 SD). X-rays excluded craniosynostosis and a later skeletal survey, at 7 years of age, did not reveal any skeletal abnormalities. At 7 years of age, she had severe ID and autistic features, her OFC was 42 cm (−7.3 SD), height 102 cm (−4.2 SD). She had microphthalmia and was blind. At 20 years of age, her OFC was 47 cm (−5.6 SD), height 144 cm (−3.8 SD), small hands and feet (shoe size: Eur 34/US 2). She has aggressive outbursts, but milder than her brother. She has no speech, but can understand some phrases. She plays a couple of simple melodies with one finger on the piano, and is able to sing along reaching the correct tone in the melody. She easily gets confused and anxious, and refuses to be held for medical investigation.
Family 3
The proband presented at age 3 years with ID/DD, attention deficit hyperactivity disorder (ADHD), strabismus and microcephaly. She was the product of a first-cousin consanguineous marriage from Egyptian parents residing in Cairo. She displayed typical gross and fine motor skills but absent language skills. Social skills were delayed by 1.5 years. Apart from one episode of seizures at 15 days of life during an intercurrent fever, there were no other neurological or general physical deformities. Head circumference at 5 years of age was 41.5 cm (−6.4 SD), and was reported to be reduced at birth, but measurements were not available. Brain MRI has shown a predominantly frontal lobe simplified gyral pattern without other major structural anomalies noted. The second child from this family was noted to be microcephalic at birth with head circumference 30 cm, and showed a similar uneventful clinical course marked by delayed development. Brain MRI at 1 year of age showed simplified gyral pattern, frontal cerebral atrophy, cerebellar hypoplasia, and partial agenesis of the corpus callosum. Head circumference at 2 8/12 years of age was 39.5 cm (−5.6 SD).
Family 4
In a consanguineous family originating from Iran, two pregnancies of a first-cousin couple were interrupted due to severe fetal malformations. Both fetuses were male and the first was aborted at 15 weeks due to severe microcephaly detected on ultrasound. Further examination demonstrated narrowing of the cranial sutures suggestive of craniosynostosis, but no other anomalies were detected. The second pregnancy was terminated at 17 weeks after detection of severe microcephaly. General growth was normal with an estimated weight of 176 g (40th centile), and there were no brain or cardiac malformations.
The successful gathering of information of affected individuals with similar phenotypes and likely disease-causing variants in the candidate causative gene was possible due to GeneMatcher (https://genematcher.org/) [16].
Variant detection
Family 1
The laboratory and bioinformatics analysis for the variant identification was performed as previously described [17]. The study was approved by the Bioethics Committee of the University Hospitals of Geneva (Protocol number: CER 11-036).
Family 2
Whole-exome sequencing (WES) was performed on the proband at the Human Genome sequencing center (HGSC) at Baylor College of Medicine through the Baylor-Hopkins Center for Mendelian Genomics initiative. Using 1ug of DNA an Illumina paired-end pre-capture library was constructed as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Pre-capture libraries were pooled into 4-plex library pools and then hybridized in solution to the HGSC-designed Core capture reagent [18] (52 Mb, NimbleGen) or 6-plex library pools used the custom VCRome 2.1 capture reagent1 (42 Mb, NimbleGen). With a sequencing yield of 9.1 Gb, the sample achieved 91% of the targeted exome bases covered to a depth of 20× or greater. Illumina sequence analysis was performed using the HGSC Mercury analysis pipeline [19, 20] (https://www.hgsc.bcm.edu/software/mercury). The clinical SNP array of the proband, parents, and two unaffected siblings was performed with Affymetrix Genome-Wide Human SNP Array 6.0 according to manufacturer’s protocol (Thermo Fisher Scientific Inc., CA, USA).
Research protocols were approved via institutional research boards (H-29697- Genome Sequencing to Elucidate the Causes and Mechanisms of Mendelian Genetic Disorders) and regional ethics committee in keeping with national guidelines, and the molecular analyses in the family were performed in accordance with the Norwegian National Biotechnology Act. All legal guardians, parents, and close family members provided written informed consent for study participation for themselves and their affected relatives as well as for publication of clinical information, molecular findings, and photographs.
Family 3
WES was performed on the proband, with >96% of the exome covered at >12×. GATK best practices pipeline was used for SNP and INDEL variant identification and XHMM was used for structural variant detection. Homozygous variant prioritization included MAF < 0.001 in a cohort of over 3000 Egyptians, and GERP score > 4.0. The study was approved by the Institutional Review Board (IRB) of UC San Diego (140028). Written informed consent was obtained from all participants.
Family 4
WES was performed on the DNA from the aborted second fetus. No DNA was available from the first affected pregnancy. On the assumption that the disease follows an autosomal recessive inheritance in the family as well as presence of consanguinity, we prioritized the homozygous potentially functional variants residing within the runs of homozygosity. These variants were screened through publicly available population databases and our in-house database generated for frequency in human population. We excluded synonymous variants, intronic variants (>5 bp from exon boundaries) and common variants (minor allele frequency, >0.001%).
Estimating potential pathogenic variants and their cumulative frequency in gnomAD
In gnomAD (http://gnomad.broadinstitute.org/), 860 missense and predicted LoF variants are reported in KIF14. Potential pathogenic variants have been selected based on a two-step approach: (i) variants with MAF higher than 0.02 and homozygous variants have been excluded, (ii) among the remaining LoF variants and missense variants predicted pathogenic by SIFT (http://sift.jcvi.org/) [21], PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) [22], and MutationTaster (http://www.mutationtaster.org/) [23] have been designated as potentially pathogenic.
Results
Variant identification
Family 1
After a combination of homozygosity mapping and exome sequencing using CATCH [24], we identified two potentially pathogenic variants, that have been confirmed through Sanger sequencing and the correct segregation in the family was verified. The first was in KIF14 NM_014875.2:c.2522C>T:p.(Ser841Phe), (dbSNP142: rs139385693). The second was in PTPRC (MIM: 151460), NM_002838.4:c.3416C>T:p.(Ser1137Phe), (dbSNP142: rs549131091). Both variants are present in ExAC in the African population, the first twice with a MAF of 0.0001924 and the second once with a MAF of 9.705e−05. Between the two, KIF14 was considered the main candidate since homozygous or compound heterozygous pathogenic PTPRC variants are responsible for a severe combined immunodeficiency (MIM: 608971) that presents at the first 2–3 months of life and that the patients of family 1 did not have.
Family 2
Clinical SNP array in this family had previously revealed several homozygous loci in the proband, not shared by two of the unaffected siblings. WES of the proband’s (Fig. 2, individual F2-IV.2) sample revealed 35 homozygous variants with ESP MAF less than 0.01, and 8 of them were not seen in any in-house or publicly available variant frequency database. Seven of these non-X-linked variants were located at sites conserved across species, and the two most interesting ones were the KIF14 variant c.246del (NM_014875.2), and a missense variant in INPP5A: c.920A>C (NM_005539.4). INPP5A is an enzyme expressed in the brain and the gene is homologous to INPP5B and to OCRL, the gene responsible for Lowe oculocerebrorenal syndrome (OMIM#309000). Familial study demonstrated that only KIF14 segregated correctly in the family.

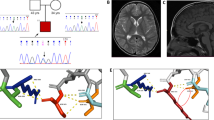
Model of human KIF14 motor domain with Gly459’s position on the L5 loop indicated with red color. ADP is presented with atom and surface representation. The hydrogen bond between Gly459 and Ser454 is indicated as cyan line
Family 3
Homozygous variants in four genes (NYNRIN, ZNF746, PXK, and KIF14) were predicted to be pathogenic. Only KIF14 NM_014875.2:c.1375G>A:p.(Gly459Arg) segregated with the disease in both affected siblings upon Sanger sequence confirmation, and occurred within a 6 Mb block of homozygosity. This variant has not been reported in either gnomAD or 1000 Genomes.
Family 4
The filtered WES data narrowed down the variants to a homozygous single nucleotide deletion in exon 29 of KIF14 (c.4432delA; p.(Ser1478fs)) located within a ~17.8 Mb region of homozygosity on chrq3-q35.3. The variant was never observed in any public variant database and it was predicted to delete a highly conserved functionally important C-terminal domain of the protein. The variant was validated by Sanger sequencing and the parents and the only healthy sibling were all heterozygous. No other pathogenic/likely pathogenic variant were identified in the currently known monogenic disease-causing genes in the WES data.
The relative positions of all the identified variants are shown in Fig. 2. All identified variants have been submitted to LOVD (https://databases.lovd.nl) with submissions ID 00132082 to 00132085.
Modeling of the missense variants
The KIF14 kinesin superfamily of microtubule-associated molecular motor proteins have an important role in intracellular transport and cell division. The motor domain of KIF14 is in the center of the protein (aa 358–701), whereas the «forkhead-associated» (FHA) domain that recognizes the phosphothreonine, is located towards the C-terminal region (aa 825–891). The two missense variants p.Gly459Arg and p.Ser841Phe are located within the motor domain and FHA domain, respectively. There is no full-length structure of KIF14 available, but there are experimental structures of single domains for close homologs of human KIF14 which were used to create homology models.
The Gly459 residue is located within the KIF14 motor domain that was modeled based on mouse KIF14 motor domain structure (4OQZ PDB) [25], which shares 83% sequence identity with the human motor domain. Gly459 is located on the L5 loop, close to the ATP/ADP binding pocket. It has a structural role in stabilizing the loop structure, as it forms hydrogen bond with Ser454 on the preceding alpha helix. In Fig. 2, the hydrogen bond is indicated as cyan line. p.(Gly459Arg) variant is most probably changing the structure of the L5 loop and may influence the ATP/ADP binding and even microtubules binding, as it is reported for variants in this region for other kinesins [26].
The Ser841 residue is in the FHA domain. It was modeled based on the 4EGX structure of CC1/FHA domain of kinesin-3, with 35% of sequence identity between both sequences. In the kinesin-3, the coiled-coil domain and FHA domain are involved in the dimerization of kinesin and regulation of motor function activity [27]. KIF14 domains are organized as in kinesin-3, which might indicate a similar role in dimerization and regulatory function of the first coiled-coil region and FHA domain. In our model of KIF14, FHA domain Ser841 residue’s side chain forms a hydrogen bond with Glu814, stabilizing the FHA domain structure (Fig. 3). Upon change from Ser841 to Phe (c.2522C>T:p.(Ser841Phe)), formation of this hydrogen bond is not possible and the stability of the FHA domain might be compromised. The change in the structure of FHA domain occurring as a consequence of the Ser841Phe variant might influence assumed FHA-mediated dimerization and this way perturb the activity of the motor domain.

Model of homodimeric structure of FHA domain of the human KIF14 protein. One domain presented in coral (left-side), second one presented in white (right-side). Side chain of residue Ser841 forming a hydrogen bond with Glu814, stabilizing the FHA structure is shown
Potential pathogenic variants in gnomAD
There are 64 predicted LoF variants for a total of 137 alleles. There are 759 missense variants after the selection process described, 170 of which are predicted to be pathogenic by all three software programs for a total of 637 alleles. Combined these two groups of variants have a cumulative MAF of 0.00279, which translates that ~1/128,000 individuals would carry two predicted pathogenic variants. This calculation represents a very crude estimation as it does not consider specific population frequencies, the possibility that two variants are in cis configuration, and is based on in silico pathogenicity prediction algorithms.
Discussion
We present four families with non-synonymous predicted loss-of-function (LoF) homozygous variants in KIF14. Table 2 presents a summary of the phenotypic findings in each one of the affected individuals along with the findings reported by FIlges et al. [15]. All patients have been identified by combining homozygosity mapping and exome sequencing. Among the pregnancies that reached term, 5/6 patients had severe ID/DD with or without autistic features and attention deficit hyperactivity disorder (ADHD), and the patients have either microcephaly (5/7) or occipital frontal circumference (OFC) at the lowest normal values (1/7) according to WHO criteria. For the majority of the additional features described in the patients there is none that is present in all of them leading to related but not identical phenotypes. As such, 5 of 7 are presenting symptoms from the ocular system, ranging from strabismus to microphthalmia, 3 of 7 presented with hypotonia, at least at the beginning of the life, 5 of 7 had either no speech or speech delay, and 3 of 7 had delay in gross motor development. Overall, 3 of 7 had short stature. There is no apparent common facial gestalt, brain imaging showed specific pathologic findings in 3 patients; and one patient had foot deformities and frequent respiratory infections. A possible explanation of the additional features may be the fact that all the patients come from consanguineous families and the possibility of a second simultaneous disorder cannot be excluded.
The patients in families 1–3 show a spectrum that seems to be less severe than the ones reported by Filges et al. [15], however, the affected fetuses in family 4 may closer resemble Filges’ report, but none has overt genitourinary signs and symptoms. In comparison with the Filges et al. [15] report that describes a lethal fetal disorder with severe brain growth restriction and IUGR due to frameshift variant, our patients represent a contiunuum towards somewhat milder phenotypes. The siblings in family 1 with a missense variant in exon 14 have severe ID and no speech, but normal head circumference, and the siblings in family 3, also with missense variant, had microcephaly but less severe ID. The patients in family 2 with the early frameshift variant have severe microcephaly with prenatal onset, as well as complete blindness with optic nerve hypoplasia and microphthalmia. In family 4, where the other frameshift variant was detected, both fetuses had been terminated at gestational age week 15th and 17th because of severe microcephaly.
The variants detected in our patients span the entire gene (from exon 2 to exon 29) and are both missense and predicted LoF. The small number of patients does not allow for definitive phenotype–genotype correlations, but in the two families with the missense variants we observe relatively milder phenotypes. In the first family, microcephaly is not observed while in the third family one of the children carrying the KIF14 variants, despite microcephaly and brain abnormalities, does not have severe developmental delay at age 2½ years of age. These observations suggest that the missense variants reported may be hypomorphs leading to less severe phenotypes. More cases are needed to study the potential phenotypic correlation with the KIF14 pathogenic variants.
A knockout mouse model has also been recently published with features similar to those described in the patients (microcephaly, slender optic nerves, small body size) [14]; these mice develop severe ataxia, and die shortly after birth, in contrast to the reported patients. No hypomyelination was identified in our patients, contrary to what was described in the mouse model.
In ExAC (http://exac.broadinstitute.org/) [28], there seems to be no constraint on the gene, neither for the missense (z = −1.18) nor for the LoF variants (pLI = 0). These metrics are compatible with genes that follow autosomal recessive inheritance. In the GTEx portal (www.gtexportal.org), the highest expression is seen in testis, esophagus, and small intestine with minimal expression in brain. On the contrary, in MGI (http://www.informatics.jax.org) expression is observed in the eye before and after birth (the eye is not interrogated in GTEx).
The KIF family of genes have already been linked to several monogenic disorders the majority of which are characterized by intellectual disability or other neurologic symptoms with various other systems affected. (Table 1). Filges et al. [15] suggested the human autosomal recessive KIF14 related disease to be a fetal lethal ciliopathy. Other kinesins may cause ciliopathy, such as Joubert syndrome in KIF7 related disease (OMIM#200990) [29]. Ciliar intraflagellar transport cargoes are transported along axonemal microtubules by kinesin and dynein motors; however, the link between ciliopathies and kinesin proteins is not yet clearly clarified for all kinesin proteins [30].
For KIF14, its function as mitotic motor protein at the spindle poles may be the cause of the neurological phenotypes observed. There are also KIF genes (KIF21A and KIF22) that are not causing neurologic phenotypes. There seems to be no preferred mode of inheritance as autosomal dominant, recessive, and X-linked modes are documented.
Primary microcephaly (MCPH) is most often caused by biallelic LoF variants in a host of genes encoding proteins functioning in cytokinesis. Most MCPH proteins show predominant localization to the centrosome and spindle pole during the cytokinesis stage of cell division [31], and mutant cells show defective spindle orientation, and reduced centrosome integrity, and reduced fidelity of chromosome distribution to the daughter cells. KIF14 is a mitotic motor protein that is required for spindle localization of the citron rho-interacting kinase, CIT, also mutated in microcephaly (OMIM#617090). At least two mechanisms have been proposed for the reduced number of neurons that results from these variants: (1) Depletion of stem cell progenitors due to a shift in balance toward asymmetric cell division and (2) Genotoxic stress due to slowed or inaccurate chromosome segregation [32]. The latter was proven to be relevant in several microcephaly models, as removal of p53, which allows cell division to continue in the presence of genotoxic stress, rescued the severe neuronal loss [33].
In addition to the focus of MCPH proteins at the spindle poles, there is attention now on their role at the midbody, which is a specialized protein complex regulating the final separation of the two daughter cells at the completion of cytokinesis [34]. KIF14 now joins ASPM and CIT as MCPH genes whose proteins localize to the midbody function. Importantly, KIF14 is required for CIT localization to the midbody. Moreover, variants affecting the protein’s function in either result in the highly specific finding of bi-nucleated daughter cells, as a reflection of failed midbody function [15, 35, 36]. These cells show similar vulnerability to genotoxic stress, and increased cellular apoptosis, which probably accounts for the severe reduction in brain size [33, 37].
The data presented here suggest that biallelic variants affecting the function of KIF14’s result in a wide phenotype spectrum involving intellectual disability, microcephaly, and short stature.
References
Marx A, Hoenger A, Mandelkow E. Structures of kinesin motor proteins. Cell Motil Cytoskelet. 2009;66:958–66.
Verhey KJ, Kaul N, Soppina V. Kinesin assembly and movement in cells. Annu Rev Biophys. 2011;40:267–88.
Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;317:73–75.
Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50.
Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–96.
Cross RA, McAinsh A. Prime movers: the mechanochemistry of mitotic kinesins. Nat Rev Mol Cell Biol. 2014;15:257–71.
Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527–39.
Corson TW, Huang A, Tsao MS, Gallie BL. KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene. 2005;24:4741–53.
Corson TW, Zhu CQ, Lau SK, Shepherd FA, Tsao MS, Gallie BL. KIF14 messenger RNA expression is independently prognostic for outcome in lung cancer. Clin Cancer Res. 2007;13:3229–34.
Theriault BL, Pajovic S, Bernardini MQ, Shaw PA, Gallie BL. Kinesin family member 14: an independent prognostic marker and potential therapeutic target for ovarian cancer. Int J Cancer. 2012;130:1844–54.
Hung PF, Hong TM, Hsu YC, et al. The motor protein KIF14 inhibits tumor growth and cancer metastasis in lung adenocarcinoma. PLoS ONE. 2013;8:e61664.
Singel SM, Cornelius C, Batten K, et al. A targeted RNAi screen of the breast cancer genome identifies KIF14 and TLN1 as genes that modulate docetaxel chemosensitivity in triple-negative breast cancer. Clin Cancer Res. 2013;19:2061–70.
Scott EM, Halees A, Itan Y, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6.
Fujikura K, Setsu T, Tanigaki K, et al. Kif14 mutation causes severe brain malformation and hypomyelination. PLoS ONE. 2013;8:e53490.
Filges I, Nosova E, Bruder E, et al. Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clin Genet. 2014;86:220–8.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Makrythanasis P, Nelis M, Santoni FA, et al. Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat. 2014;35:1203–10.
Bainbridge MN, Wang M, Wu Y, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68.
Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8.
Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinforma. 2014;15:30.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Santoni FA, Makrythanasis P, Antonarakis SE. CATCHing putative causative variants in consanguineous families. BMC Bioinformatics. 2015;16:310.
Arora K, Talje L, Asenjo AB, et al. KIF14 binds tightly to microtubules and adopts a rigor-like conformation. J Mol Biol. 2014;426:2997–3015.
Behnke-Parks WM, Vendome J, Honig B, Maliga Z, Moores C, Rosenfeld SS. Loop L5 acts as a conformational latch in the mitotic kinesin Eg5. J Biol Chem. 2011;286:5242–53.
Huo L, Yue Y, Ren J, et al. The CC1-FHA tandem as a central hub for controlling the dimerization and activation of kinesin-3 KIF1A. Structure. 2012;20:1550–61.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Liem KF Jr., He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci USA. 2009;106:13377–82.
Verhey KJ, Dishinger J, Kee HL. Kinesin motors and primary cilia. Biochem Soc Trans. 2011;39:1120–5.
Faheem M, Naseer MI, Rasool M, et al. Molecular genetics of human primary microcephaly: an overview. BMC Med Genome. 2015;8(Suppl 1):S4
Di Cunto F, Imarisio S, Hirsch E, et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–27.
Bianchi FT, Tocco C, Pallavicini G, et al. Citron kinase deficiency leads to chromosomal instability and TP53-sensitive microcephaly. Cell Rep. 2017;18:1674–86.
Paramasivam M, Chang YJ, LoTurco JJ. ASPM and citron kinase co-localize to the midbody ring during cytokinesis. Cell Cycle. 2007;6:1605–12.
LoTurco JJ, Sarkisian MR, Cosker L, Bai J. Citron kinase is a regulator of mitosis and neurogenic cytokinesis in the neocortical ventricular zone. Cereb Cortex. 2003;13:588–91.
Harding BN, Moccia A, Drunat S, et al. Mutations in citron kinase cause recessive microlissencephaly with multinucleated neurons. Am J Hum Genet. 2016;99:511–20.
Li H, Bielas SL, Zaki MS, et al. Biallelic mutations in citron kinase link mitotic cytokinesis to human primary microcephaly. Am J Hum Genet. 2016;99:501–10.
Acknowledgements
We would like to thank the patients and their families. The University of Geneva project (family 1) was supported by grants from the SNF to SEA and von Meissner Foundation to PM. The Baylor-Hopkins Center for Mendelian Genomics (family 2) is supported by the US National Human Genome Research Institute/National Heart Lung and Blood Institute (UM1HG006542). The study of family 3 was supported by NIH grants P01HD070494, R01NS052455, the Simons Foundation Autism Research Initiative, Howard Hughes Medical Institute (to JGG) and the Broad Institute (UM1HG008900).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Periklis Makrythanasis, Reza Maroofian, Asbjørg Stray-Pedersen, and Damir Musaev contributed equally to this work.
Joseph G. Gleeson, James R. Lupski, Ehsan Ghayoor Karimiani, and Stylianos E. Antonarakis contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Makrythanasis, P., Maroofian, R., Stray-Pedersen, A. et al. Biallelic variants in KIF14 cause intellectual disability with microcephaly. Eur J Hum Genet 26, 330–339 (2018). https://doi.org/10.1038/s41431-017-0088-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0088-9
This article is cited by
-
TEFM variants impair mitochondrial transcription causing childhood-onset neurological disease
Nature Communications (2023)
-
KIF5C deficiency causes abnormal cortical neuronal migration, dendritic branching, and spine morphology in mice
Pediatric Research (2022)
-
Pontocerebellar hypoplasia due to bi-allelic variants in MINPP1
European Journal of Human Genetics (2021)
-
Pathogenic variants in PIDD1 lead to an autosomal recessive neurodevelopmental disorder with pachygyria and psychiatric features
European Journal of Human Genetics (2021)
-
Inhibiting microcephaly genes as alternative to microtubule targeting agents to treat brain tumors
Cell Death & Disease (2021)
