
Abstract
Whole-gene duplications and missense variants in the HUWE1 gene (NM_031407.6) have been reported in association with intellectual disability (ID). Increased gene dosage has been observed in males with non-syndromic mild to moderate ID with speech delay. Missense variants reported previously appear to be associated with severe ID in males and mild or no ID in obligate carrier females. Here, we report the largest cohort of patients with HUWE1 variants, consisting of 14 females and 7 males, with 15 different missense variants and one splice site variant. Clinical assessment identified common clinical features consisting of moderate to profound ID, delayed or absent speech, short stature with small hands and feet and facial dysmorphism consisting of a broad nasal tip, deep set eyes, epicanthic folds, short palpebral fissures, and a short philtrum. We describe for the first time that females can be severely affected, despite preferential inactivation of the affected X chromosome. Three females with the c.329 G > A p.Arg110Gln variant, present with a phenotype of mild ID, specific facial features, scoliosis and craniosynostosis, as reported previously in a single patient. In these females, the X inactivation pattern appeared skewed in favour of the affected transcript. In summary, HUWE1 missense variants may cause syndromic ID in both males and females.
Similar content being viewed by others


Introduction
The HUWE1 gene (HECT, UBA and WWE domain containing 1, E3 ubiquitin protein ligase; MIM 300697), located on Xp11.22, encodes a large protein of 4374 amino acids initially identified in oncogenesis [1]. Mouse models have indicated the important role of HUWE1 in the control of neurogenesis in the cerebral cortex via the N-Myc pathway [2,3,4]. Deletion of Huwe1 in the progenitors of the embryonic mouse brain or only in cerebellar neuron precursors and radial glia, leads to neonatal lethality [3, 4].
The association of HUWE1 variants or rearrangements with X-linked intellectual disability (XLID) is now well recognised. In 12 families with mild to moderate non-syndromic XLID, Froyen et al. identified overlapping microduplications at Xp11.22, encompassing HUWE1 and HSD17B10, ranging in size between 0.4 and 1 Mb [5, 6]. With high-resolution mapping of the 12 copy number gains, they showed that HUWE1 was the only gene in the minimal duplicated region. Moreover, a twofold increased expression of HUWE1 (but not HSD17B10) was seen in cell lines from patients with cognitive impairment [6]. In the initial report, the authors also identified three different missense variants in HUWE1 (c.8942 G > A p.(Arg2981His); c.12037 C > T p.(Arg4013Trp) and c.12559 C > T p.(Arg4187Cys)) in three families with non-syndromic moderate to profound XLID [5, 7]. Carrier females are usually described as normal or less symptomatic, which previous authors attribued to protective skewed inactivation of their affected X chromosome [6]. Isrie et al. reported the c.12037 C > T (p.(Arg4013Trp)) variant in HUWE1 in another XLID family in which two affected male patients presented with severe intellectual disability (ID), deep set eyes, downslanting palpebral fissures, tapering fingers and oedema of the hands and feet [8]. Missense variants have since been reported in patients with ID, sometimes in association with autism or schizophrenia, but clinical information about these patients are limited [9,10,11,12,13,14]. Recently, Friez et al. described a recurrent c.12928 G > C (p.Gly4310Arg) missense variant in HUWE1 in two unrelated XLID families, formerly described by Juberg and Marsidi and by Brooks [15,16,17]. In a third family, a c.12188 G > A (p.Arg4063Gln) variant was identified in two brothers [15]. In all three families, affected male patients shared similar clinical features including severe ID, with absence of speech in all but one patient, short stature, microcephaly, contractures, and dysmorphic features such as blepharophimosis, deep set eyes and a prominent nose. However the contribution of HUWE1 variants to ID in female patients is not well understood. The first female patient with a de novo c.329 G > A (p.Arg110Gln) variant in HUWE1 had learning difficulties and craniosynostosis [18]. She showed unfavourable skewing of her X inactivation pattern and expressed only the mutant allele in both lymphoblastoid cells and fibroblasts. In addition, two males with craniosynostosis and global developmental delay with de novo c.328 C > T (p.(Arg110Trp)) have been described [18,19,20], adding to the probability that p. Arg110 missense variants cause a specific phenotype.
Here, we describe the clinical and molecular findings in the largest cohort to date of 14 female and 7 male patients with HUWE1 variants, and review the current literature on HUWE1 aberrations.
Patients and methods
Patients
The 21 patients were assessed clinically by at least one of the authors. Written informed consent was obtained for genetic studies and publication of all photographs. All patients were referred for investigation of syndromic/non-syndromic developmental delay and/or ID of unknown cause. Nine patients were ascertained through the Deciphering Developmental (DDD) Study [21] (See Table 1 for associated DECIPHER ID numbers).
Four patients have been previously reported (P2, P8, P13 and P14) [18, 22, 23]. For each of them, the clinical data have been reassessed. P13 and P14 were clinically described by Verloes et al. [23] and included in our cohort because the molecular diagnosis had not yet been identified. In total, we describe 21 individuals, 14 females and 7 males, with an age range of 18 months to 31 years.
Genetic studies
Blood samples from individuals and their parents were collected for trio whole-exome sequencing (WES: P3–P8, P9–P15, P17–P19), trio WES with filtering for ID genes (ID-panel: P1 and P16) or X chromosome exome sequencing (XES: P20–P21) (Table 1).
The molecular methods and bioinformatic pipeline for WES used in the DDD Study have been previously described [21]. All variants in the HUWE1 gene (NM_031407.6) were confirmed by Sanger sequencing. They were interpreted and classified according to the ACMG 2015 Guidelines [24]. Variants were submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). ClinVar accession numbers are listed in Supplementary Table S1.
For the X chromosome inactivation (XCI) assay, we used the polymorphic CAGn repeat within the human AR gene to assess the relative methylation status of both chromosomes after methylation-sensitive restriction enzyme digest to assess the XCI patterns as previously described [25]. X inactivation was considered skewed (non-random) if the ratio of the two alleles exceeded 80:20 (we refer to ‘extreme skewing’ where the ratio exceeds 90:10) [26]. XCI profiles were performed in peripheral blood leucocytes from individuals P1, P3–P5, P7, P9–P10, P15–19, and in the mother and grandmother of P20 and P21 (Table 1 and Fig. 1). The analysis was also performed on a buccal swab sample of P1, P11, P15–P17, and in cultured fibroblasts in P1, P4, P5, P7, P15 and P16.

Pedigrees of families with maternally inherited HUWE1 variants. Red arrow indicates the proband; +, hemizygous status; +/-, heterozygous status, -/-, wt. Numbers in brackets indicate the XCI pattern in female carriers. (Color figure online)
RNA isolation and sequencing of cDNA
Total RNA was isolated from fibroblasts from patient P1, P4, P5, P7, P15, P16 and from blood samples obtained from P1, P4, P11, P17 and controls, by using the RNeasy Mini Kit (QIAGEN) for fibroblasts and PAXgene Blood RNA Kit (QIAGEN) for blood samples, following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesised following the manufacturer’s protocols. The different variants were amplified by PCR from the patient’s cDNA (amplified from total RNA by reverse transcriptase-polymerase chain reaction (PCR)) from fibroblasts and blood by primers spanning the different variants. The primer sequences can be obtained by request. PCR amplification and the sequencing reactions were performed by standard methods. RNA studies of patient P2 were previously described [18].
Results
HUWE1 variants
We report 21 patients with 16 different HUWE1 variants (Table 1, Fig. 1). In 17 cases, the HUWE1 variants were de novo. In two unrelated families (Fig. 1), two male patients both had a maternally inherited mutation. With the exception of a de novo splice site variant identified in P5, all the missense variants predict substitutions that affect highly conserved amino acids. None of the variants were reported in control databases (Exome Variant Server, 1000Genomes, HapMap, Exome Aggregation Consortium), whereas the c.9208 C > T p.(Arg3070Cys) variant was reported in the ClinVar database (P11). This variant is recurrent, found in P11 and P12. The recurrent c.329 G > A p.Arg110Gln variant found in P1 and P3 was identified in a previously reported patient [18] included in our cohort (P2) with updated and detailed clinical findings. The 16 different variants are located in 5 out of 6 different functional domains of the protein (Fig. 2), but with an overrepresentation (7/16) in the catalytic HECT domain.

Schematic representation of HUWE1 protein with identified variants. (Refseq: NM_031407.6, NCBI Protein Reference Sequence: NP_113584.3). The amino-acid borders of the described HUWE1 domains are noted. Previously published variants are shown on the left and variants in the present cohort on the right. Only rare non-synonymous and splice site variants are presented. Male patients are in blue and female in red. Number in parenthesis indicates recurrent variants describing number of families reported for the variant. *Females with schizophrenia as indication for WES with de novo HUWE1 missense changes. DUF domain of unknown function; UBA ubiquitin-associated; WWE tryptophan tryptophan glutamate; HECT homologous to the E6-AP Carboxyl Terminus. (Color figure online)
XCI and cDNA results
The majority of female patients tested for XCI (13/14) had a skewed X inactivation ratio (>80%) except in P9 (Table 1, Fig. 3). P7 was non-informative, but cDNA sequencing indicated extreme skewing, as no mutant allele was expressed in RNA. Unaffected heterozygous carrier females (mothers, aunts and grandmother of P20 and P21) also presented with skewed XCI (Fig. 1). XCI was tested in DNA from different tissues (blood, buccal swab and cultured fibroblasts) in P1, P4, P5, P15, P16 and P17 (see Table 1 and Fig. 3). In P1, the XCI pattern was consistent. In P4 and P5, the pattern differed. P4 had random inactivation (60/40) in blood but skewed in fibroblasts (84/16). In contrast, P5 had an extremely skewed pattern in blood (95/5), with a random pattern in the fibroblasts (72/28). In P15, the XCI pattern was concordant between blood and buccal swab (100/0), whereas it was random (60/40) in the cultured fibroblasts. In P16, the pattern differed, from 100/0 in blood, 92/8 in cultured fibroblasts and 69/31 (random) from buccal swab. In P17, XCI was skewed (91/9) in the blood and random (74/26) in buccal swab.

X inactivation patterns in female patients in varying tissues and associated RNA expression analysis. cDNA sequencing was used to determine which allele was preferentially expressed; the green colour indicated the expression pattern of the wild-type allele and the red colour indicated the expression pattern of the mutated allele. The numbers superimposed on bars represent XCI in the specified tissue. *P2 analysed by Taylor et al. In P7, XCI was non-informative due to homozygous AR-alleles, but wt-allele exclusively expressed in blood. In P11, cDNA analysis was performed in one tissue demonstrating preferential expression of the wild-type allele. This was assumed to be true for both tissues. (Color figure online)
To determine whether the wild-type or the mutated allele was preferentially expressed, we performed cDNA sequencing (see Fig. 3). In P1, the mutated allele was preferentially expressed in the blood and exclusively expressed in the fibroblasts. A reversed pattern was demonstrated in P4 with preferential expression of the mutated allele in fibroblasts vs. wild-type in blood. In P5, the splice site variant c.567 + 1 G > C occurred in a splice-donor site in intron 8. cDNA analysis from fibroblasts confirmed that the variant resulted in an abnormal but expressed RNA product with skipping of exon 8 (NG_016261.2), leading to an in-frame deletion of 21 amino acids. cDNA sequencing could not determine which allele was preferentially inactivated in fibroblasts (XCI 72/28 in fibroblasts). In P7, P15 and P16, the wild-type allele was preferentially or exclusively expressed in the fibroblasts, as was seen in P11 and P17 in the blood. In P11, the XCI was only analysed in buccal swab, where it showed a random pattern (66/34), but RNA analysis from blood showed exclusive expression of the wild-type allele, indicating an extremely skewed XCI in blood.
Clinical features
All individuals presented with ID (Table 1). In all, 16/19 had severe to profound ID, whereas three were mildly affected (P1, P2, P9) and P3 is too young to be assessed (P13 died at 9 months from status epilepticus). The majority had global developmental delay (20/21). Hypotonia was present in 14/20. The mean age of walking was 2 ½ years while three patients never achieved autonomous walking (P4, P5 and P16) and one patient lost the use of the walk at 7 years due to contractures of knees (P15). In total, 17/19 had speech delay, with absence of speech (or <5 words) in 13/19 patients. Microcephaly (−2.5 to −7 SD) occurred in 11/21, and was often postnatal in onset. Seizures were reported in 7/18 patients with onset between 9 months and 13 years.
Brain MRI was normal in nine patients. Thin corpus callosum was noted in two and enlarged cerebral ventricles in four. Autistic features were reported in seven patients as well as hand stereotypies (8/17).
Low birth weight was noted in four patients and postnatal short stature (−2.5 to −6 SD) reported in 15/21. Skeletal anomalies (Fig. 4b) included small hands and feet (12/21), overlapping toes (9/21), craniosynostosis (2/21), scoliosis (3/21) and contractures of knees (5/17).

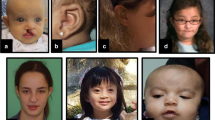
Facial and limb features of individuals with HUWE1 variants, frontal and lateral views. a Facial features of all but two patients in the cohort. Patient numbers correspond to those in the text and tables. Detailed description of facial phenotype can be found in Table 1. Note the particular phenotype in P1 to P3 (carrying the same de novo c.329 G > A p.Arg110Gln variant) consisting of a flat face, prominent eyes and a small nose, compared with other patients (P4–P20) who present with deep set eyes, epicanthic folds, blepharophimosis, broad nasal tip and thin upper lip. The facial shape seems to evolve with time, from round face with full cheeks to long face, as seen in P6, P19 and P20. b Skeletal features include small hands, short distal phalanges and short nails in P1–P3, clinodactyly of 5th fingers, small nails, short metatarsals and 3rd–5th toes in P1 and P2, 2–3 syndactyly in P1 and P3. Tapering of fingers and puffy hands are shown in P8, and overlapping toes in P16 and P17. (Color figure online)
Less common features included hyperpilosity (4/21), hearing loss (4/18), sleep disorder (5/21), hyperactivity (4/16), cryptorchidism (3/8) and hypertonia of lower limbs (2/21). Constipation occurred in four patients and feeding difficulties in two.
Dysmorphic facial features (Fig. 4a) frequently reported included a long face (11/21), a broad nasal tip (19/21), a short philtrum (10/21) with thin upper lip (16/21) and full lower lip (8/21), low set ears (7/19) or posteriorly rotated ears (8/19). The eye anomalies were most highly characteristic with deep set eyes (15/21), epicanthic folds (14/21) and blepharophimosis (12/21). Strabismus was reported in 13/20, hypermetropia ± astigmatism in 9/18 and retinopathy in 3/13.
Discussion
HUWE1 encodes an E3 ubiquitin ligase ubiquitously expressed and important in neuronal development [2,3,4]. In human EBV cell lines, there is no difference between HUWE1 expression in males and females, suggesting the gene does not escape X inactivation [6]. Female embryonic stem cell studies confirm that HUWE1 is subject to XCI but at a later stage of differentiation [27]. HUWE1 is a dosage-sensitive gene initially identified in non-syndromic XLID families with microduplication Xp11.22 encompassing HUWE1 [5, 6]. In a Drosophila melanogaster model, a twofold overexpression of Huwe1 was not associated with structural brain anomalies, however, abnormal axon branching of dorsal cluster neurons through the Wnt/β-catenin pathway was demonstrated [28].
We report here on the largest cohort of individuals with HUWE1 variants, consisting of 21 patients with 16 different variants. All but one were missense, a pattern that concurs with all previously reported patients [5, 8,9,10,11,12, 15, 18,19,20, 22, 29].
HUWE1 is a large gene, but almost devoid of truncating variants in the ExAC database. The calculated probability of loss-of-function intolerance (pLI) is 1.0, and genes with pLI ≥ 0.9 are considered extremely loss of function intolerant. Missense variants in HUWE1 are extremely rare, compared with the expected, and the ExAC database has calculated a z-score of 8.75, which is highly significant. This is also reflected in our series, where all affected sequences are highly conserved on both nucleotide and amino acid level, along with CADD-scores above 20 (Supplementary Table S1).
The physiopathological mechanisms responsible for alteration of HUWE1 function are still uncertain. Both duplications and missense variants have been associated with ID suggesting that increased (Gain of Function) or decreased loss of function function of HUWE1 could cause the disease. Whole HUWE1 gene deletions and truncating mutations have not been reported in males so far. Furthermore, the high rate of perinatal lethality in Huwe1 knockout mice would suggest that complete loss-of-function is likely to be incompatible with embryonic development [2, 4, 6]. Moreover, functional studies on lymphoblastoid cell lines derived from affected male patients have already suggested that missense variants in HUWE1 could affect its function through different mechanisms [15]. Indeed, for a p.Gly4310Arg affected male patient, the authors found reduced protein levels of HUWE1 compared with healthy relative’s cells, as well as accumulation of Mcl-1 and p53, downstream targets of HUWE1. But on the other hand, for an affected patient with p.Arg4063Gln, increased HUWE1 protein levels were observed with consequently reduced levels of p53 substrates. For others, missense variants have led to increased auto-ubiquitination of HUWE1 in vitro [30]. In our cohort, the consequences of the missense and the splice site variants are not yet understood. We hypothesise that the p.Arg100Gln variant responsible for a specific phenotype could result from a different molecular mechanism compared with the other missense variants reported. Additional functional studies in our series will be necessary to further understand the impact of these variants. These studies would need to compare HUWE1 and target proteins levels using available patients’ fibroblasts, and to study HUWE1 protein stability with overexpression of mutants in cellular models.
Prior to this work, 26 male patients from 9 unrelated families and 11 sporadic male and female individuals with missense HUWE1 variants have been reported in the literature [5, 8,9,10,11,12, 15, 18, 19, 22, 29]. We have excluded from our discussion six sporadic patients whose descriptions lack accurate phenotypic information [9,10,11,12, 29]. All 31 published males have moderate to profound ID, which is a consistent feature that we found in our study, confirming that HUWE1 variants are associated with significant ID. Absent or limited speech is reported in 75 % of published patients and in 89 % of our cohort. Postnatal short stature occurred in 70% of patients in the literature, with the same rate in our series (71%). We highlight recurrent facial dysmorphism in our patients and those previously reported, consisting of a long face, a short nose with a broad nasal tip, deep set eyes with epicanthic folds, blepharophimosis, thin upper lip and full lower lip. However, the dysmorphic features do not present as a specifically recognisable gestalt. Skeletal anomalies consist of small hands and feet (57%), overlapping toes (42%) and contractures of the knees and other large joints. This last characteristic was previously reported [5, 7, 15,16,17]. Ophthalmologic anomalies must be screened for, in particular strabismus (65% of all patients), hypermetropia and/or astigmatism (50%).
Of note, five patients have variants affecting the same residue p.Arg110 of HUWE1. Three were female patients from this cohort with the c.329 G > A p.Arg110Gln substitution (P1-P3), and the other two were male patients with c.328 C > T p.(Arg110Trp) variant who have previously been reported in the literature [18,19,20]. Both male p.(Arg110Trp) patients presented with craniosynostosis, ID and Chiari malformation. One had the additional features of scoliosis, shortening of digits, small toes with 4–5 syndactyly and facial dysmorphism (flat midface, downslanting palpebral fissures and low set ears), which is different to the dysmorphism we have observed in our cohort [18, 20]. Of the three female patients with p.Arg110Gln, two have craniosynostosis (multilacunar in P2, lacunar skull defects and unilateral coronal and metopic craniosynostosis in P3). P1 and P2 (p.Arg110Gln) have similar facial dysmorphism with a high forehead, downslanting palpebral fissures, prominent eyes, flat midface, small nose with hypoplastic nares, nasal and high-pitched voice, and oligodontia. They have short distal phalanges with small nails. Short stature and scoliosis is seen in P1. P1 and P2 have mild ID, while P3 is too young to assess but showing global developmental delay. P4 has a severe neurologic phenotype, whereas the facial phenotype is not typical for the rest of the cohort, but might resemble P1–P3. She also shares Chiari malformation in common with the p.Arg110Gln patients. In summary, we note that some dysmorphic features are different in patients P1–P4 compared with patients with different variants suggesting a possible specific phenotype.
Robust genotype–phenotype correlations are difficult to establish at this point because of low numbers. In accordance with previously reported cases, the 16 different variants in our cohort are located in five out of six different functional domains of the protein (Fig. 2). However, their location does not suggest an obvious explanation for the phenotypic variability in the patients. We noted an overrepresentation of variants (7/16) in the catalytic HECT domain but could not detect a specific phenotype in these patients. There was no significant difference between the female and male groups either overall or at specific domains.
We also describe two unrelated females with a de novo c.9208 C > T p.(Arg3070Cys) variant (P10 and P11). P11 was previously reported in the ClinVar database, but without clinical information. Although both females are phenotypically similar to the wider cohort, P10 had severe ID and absence of speech, whereas P11 had moderate ID with late-normal motor milestones. In the literature, we found four recurrent variants (c.8942 G > A p.(Arg2981His), c.12037 C > T p.(Arg4013Trp), c.12559 C > T p.(Arg4187Cys) and c.12928 G > C p.Gly4310Arg in HUWE1) [5, 8, 15]. No obvious genotype–phenotype correlation could be established apart from the recurrent p.Gly4310Arg variant reported in two families who have previously been described as Juberg–Marsidi syndrome and Brooks syndrome, respectively [16, 17]. These patients share in common severe syndromic ID with absence of speech, short stature, contractures, blepharophimosis, epicanthus and deep set eyes, a thin upper lip, cupped ears and a bulbous nose, the same dysmorphism as described in our patients.
We report for the first time a severe phenotype in females with HUWE1 variants. Previously reported carrier females were described as normal or mildly symptomatic, presumed to be due to the protective effect of skewed X inactivation of the mutated X chromosome. However, in our series, severely affected female patients also had an extremely skewed X inactivation pattern, and cDNA sequencing revealed almost exclusive expression of the normal allele (P7, P11, P15, P16 and P17). As the pattern of skewing observed in blood may not be representative of all tissues, differential X inactivation patterns in different tissues may explain the different phenotypes. The buccal swab sample (P1, P16 and P17) and cultured fibroblasts of P1 demonstrate this. Patients P1, P2 and P3 with c.329 G > A p.Arg110Gln variants all showed a specific phenotype with mild ID plus facial dysmorphism, skeletal anomalies and craniosynostosis, and all had completely skewed inactivation of their X chromosome with preferential inactivation of the wild-type X chromosome. This phenomenon has been widely described with X-linked variants [26, 31], but the cause of such apparently disadvantageous skewing is not clear. We postulate that this specific HUWE1 variant could be responsible for this specific phenotype. We confirmed by cDNA analysis that P1 preferentially expressed the mutant transcript in both leucocytes and fibroblasts. Similar findings were reported for P2 in Taylor et al. [13]. P4 with c.344 C > T p.(Ser115Phe) variant presented an atypical phenotype, and the X inactivation pattern in fibroblast from her was comparable to the pattern seen in the p.Arg110Gln-patients, with unfavourable skewing.
In conclusion, patients with variants in HUWE1 typically present with moderate to profound ID, short stature, severe speech difficulties and nonspecific but recurrent dysmorphic facial features such as deep set eyes and a broad nasal tip. In males, the majority of variants are de novo, a minority are maternally inherited from healthy female carriers demonstrating an extremely skewed pattern of X inactivation in favour of the normal X. We have identified HUWE1 variants in female patients with a severe syndromic ID phenotype. Clearly affected females have de novo variants with skewed XCI. The c.329 G > A p.Arg110Gln variants are associated with a specific phenotype including craniosynostosis, and with an unexpected XCI pattern favouring the mutated allele. It is now important to identify additional individuals with HUWE1 variants to confirm the phenotypes related to this gene, and to clarify genotype–phenotype correlations.
References
Zhong Q, Gao W, Du F, et al. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005;121:1085–95.
Zhao X, Heng JI, Guardavaccaro D, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol. 2008;10:643–53.
Zhao X, D DA, Lim WK, et al. TheN-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev Cell. 2009;17:210–21.
D’Arca D, Zhao X, Xu W, et al. Huwe1 ubiquitin ligase is essential to synchronize neuronal and glial differentiation in the developing cerebellum. Proc Natl Acad Sci USA 2010;107:5875–80.
Froyen G, Corbett M, Vandewalle J, et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet. 2008;82:432–43.
Froyen G, Belet S, Martinez F, et al. Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am J Hum Genet. 2012;91:252–64.
Turner G, Gedeon A, Mulley J. X-linked mental retardation with heterozygous expression and macrocephaly: pericentromeric gene localization. Am J Med Genet. 1994;51:575–80.
Isrie M, Froyen G, Devriendt K, et al. Sporadic male patients with intellectual disability: contribution of X-chromosome copy number variants. Eur J Med Genet. 2012;55:577–85.
Nava C, Lamari F, Heron D, et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry 2012;2:e179.
McCarthy SE, Gillis J, Kramer M, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 2014;19:652–8.
Hu H, Haas SA, Chelly J, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 2016;21:133–48.
Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014;506:179–84.
Piton A, Redin C, Mandel JL. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet. 2013;93:368–83.
Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–43.
Friez MJ, Brooks SS, Stevenson RE, et al. HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study. BMJ Open 2016;6:e009537.
Juberg RC, Marsidi I. A new form of X-linked mental retardation with growth retardation, deafness, and microgenitalism. Am J Hum Genet. 1980;32:714–22.
Brooks SS, Wisniewski K, Brown WT. New X-linked mental retardation (XLMR) syndrome with distinct facial appearance and growth retardation. Am J Med Genet. 1994;51:586–90.
Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–26.
Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–81.
Miller KA, Twigg SR, McGowan SJ et al. Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet. 2016;54(4):260–268.
Wright CF, Fitzgerald TW, Jones WD, et al. Genetic diagnosis of developmental disorders in the DDD Study: a scalable analysis of genome-wide research data. Lancet 2015;385:1305–14.
Gauthier-Vasserot A, Thauvin-Robinet C, Bruel AL, et al. Application of whole-exome sequencing to unravel the molecular basis of undiagnosed syndromic congenital neutropenia with intellectual disability. Am J Med Genet A 2017;173:62–71.
Verloes A, Bremond-Gignac D, Isidor B, et al. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A 2006;140:1285–96.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Allen RC, Zoghbi HY, Moseley AB, et al. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39.
Fieremans N, Van Esch H, Holvoet M, et al. Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum Mutat. 2016;37:804–11.
Chow JC, Ciaudo C, Fazzari MJ, et al. LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 2010;141:956–69.
Vandewalle J, Langen M, Zschatzsch M, et al. Ubiquitin ligase HUWE1 regulates axon branching through the Wnt/beta-catenin pathway in a Drosophila model for intellectual disability. PLoS ONE 2013;8:e81791.
Niranjan TS, Skinner C, May M, et al. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS ONE 2015;10:e0116454.
Sander B, Xu W, Eilers M et al. A conformational switch regulates the ubiquitin ligase HUWE1. Elife. 2017;6:e21036 https://doi.org/10.1038/s41431-017-0038-6.
Tzschach A, Grasshoff U, Beck-Woedl S, et al. Next-generation sequencing in X-linked intellectual disability. Eur J Hum Genet. 2015;23:1513–8.
Acknowledgements
We thank the patients, their families and the clinical staff worldwide for their participation. We thank the 'Fonds Marguerite-Marie Delacroix' for the research grant provided to S.M. This work was supported by the ‘Institut de Recherche Scientifique en Pathologie et Génétique’. The DDD Study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC), and the Norwegian Regional Committees for Medical and Health Research Ethics approval (2016/1909). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. Spanish patient study was supported by grant PI14/00350 (Instituto de Salud Carlos III -Acción Estratégica en Salud 2013–2016; FEDER -Fondo Europeo de Desarrollo Regional). We also would like to thank G. Matre at Haukeland University Hospital for performing all the RNA isolation and cDNA sequencing in this study. A.O.M.W. is supported by a Wellcome Senior Investigator Award (102731).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Stéphanie Moortgat, Siren Berland, Karen J. Low and Ruth A. Newbury-Ecob authors are contributed equally to this work.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moortgat, S., Berland, S., Aukrust, I. et al. HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. Eur J Hum Genet 26, 64–74 (2018). https://doi.org/10.1038/s41431-017-0038-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0038-6
This article is cited by
-
Integrated omics analyses clarifies ATRX copy number variant of uncertain significance
Journal of Human Genetics (2024)
-
Proteasome-Associated Syndromes: Updates on Genetics, Clinical Manifestations, Pathogenesis, and Treatment
Journal of Clinical Immunology (2024)
-
A study on genotypes and phenotypes of short stature caused by epigenetic modification gene variants
European Journal of Pediatrics (2024)
-
Compromised transcription-mRNA export factor THOC2 causes R-loop accumulation, DNA damage and adverse neurodevelopment
Nature Communications (2024)
-
The HAPSTR2 retrogene buffers stress signaling and resilience in mammals
Nature Communications (2023)
