
Abstract
A thermophilic bacterium Thermosporothrix hazakensis NBRC 105916 which belongs to the class Ktedonobacteria was investigated to explore its biosynthetic potential of secondary metabolites. UV-guided fractionation led to the identification of a new benzenoid metabolite designated ktedonoketone (6) and an α-diketone metabolite 2’-oxosattabacin (7) along with five known compounds. Compound 7 was previously described as a synthetic compound, but this is the first finding as a natural product. Compound 7 induced adipocyte differentiation at 10–20 μM and autophagy at 1–10 μM. Compound 6 showed weak inducing activity of adipocyte differentiation. The biosynthetic origin of hazakacin (3), an acyloin-type compound, was elucidated by 13C-labeled precursor-feeding experiments.
Similar content being viewed by others

Introduction
After the intensive screening efforts on common mesophilic microorganisms, extreme environments such as deserts and deep sea are drawing attention as a remaining niche for unexplored or underexplored microbes for drug discovery [1, 2]. Ktedonobacter racemifer was first isolated from soil in Italy. It was Gram-positive and displayed morphological similarity to actinomycetes, developing a multicellular filamentous structure with branched mycelia and grape-like spores [3]. However, its 16S rRNA gene sequence was nearest to the species in the phylum Chloroflexi (78–79% similarity to Caldilnea, Roseiflexus, and Sphaerobacter), phylogenetically apart from actinomycetes. Accordingly, a new taxon, class Ktedonobacteria was created in this phylum, currently including two additional genera Thermosporothrix and Thermogemmatispora [4, 5]. Except the first identified K. racemifer that grows around 30 °C, other Ktedonobacteria species display thermophilic characteristic with a preference to higher temperature ranging from 50 to 65 °C for optimal growth [6]. In addition to its actinomycete-like morphology, another notable feature of Ktedonobacteria is its relatively large genome size: the genome sizes are 13.7 Mbp for K. racemifer SOSP1-21T, 7.3 Mbp for T. hazakensis SK20-1T, and 5.6 Mbp for Thermogemmatispora carboxidivorans PM5T [7,8,9]. Analysis of the genome sequence of K. racemifer SOSP1-21T suggested that more than 200 genes are likely involved in secondary metabolism [7]. Meanwhile, only five metabolites are known from the class Ktedonobacteria: an unusual branched fatty acid (1) from Thermogemmatispora [10], and two acyloins, sattabacin (2) and hazakacin (3) [8], and two thiazole derivatives (4 and 5) from T. hazakensis (Fig. 1) [11].

Secondary metabolites identified from Ktedonobacteria
As a part of our continuing study on metabolic capability of thermophilic bacteria [12, 13], secondary metabolite production by T. hazakensis NBRC 105916 was investigated. In this paper, we report the isolation of ktedonoketone (6) and 2’-oxosattabacin (7) along with several known benzenoid metabolites (2, 3, 4, 8 – 11, Figs. 1, 2). Biosynthetic origin of an acyloin-type compound hazakacin (3) was also studied by 13C-labeled precursor-feeding experiments.

Metabolites isolated from Thermosporothrix hazakensis NBRC 105916
Results and discussion
The producing strain T. hazakensis NBRC 105916 was precultured in the V22 medium at 50 °C and then cultured in the A-11M production medium at 50 °C for 6 days. The whole-culture broth was extracted with 1-butanol, and the concentrated extract was fractionated by silica gel and ODS column chromatographies. Final purification was achieved by preparative HPLC to give ktedonoketone (6, 2.9 mg) and 2’-oxosattabacin (7, 3.2 mg) from 6 l of main culture.
Ktedonoketone (6) was obtained as an optically active yellow oil. HRESITOFMS analysis gave a molecular formula of C14H16O2, which was consistent with the 1H and 13C NMR data. The IR spectrum displayed absorption bands for hydroxy and carbonyl groups (3378 and 1703 cm−1). Analysis of 13C NMR and HSQC spectral data confirmed the presence of 11 carbons assignable to two downfield sp2 carbons (δC 210.2 and 172.3), seven sp2 carbons (five are proton-bearing), one oxygenated sp3 carbon, two sp3 methylenes, and two methyl carbons (Table 1). The 1H NMR spectrum showed a typical resonance pattern for a phenyl group at δH 7.26 (2H, d), 7.37 (2H, t), and 7.32 (1H, t). The sp2 carbon at δC 133.0 (C-6) was placed in this aromatic ring by HMBC correlations from the protons at δH 7.37 (H8 and H10) to this carbon. In addition, two methyl proton signals at δH 0.94 (3H, t) and 2.20 (3H, s), geminal methylene proton signals at δH 2.66 (1H, d) and 2.86 (1H, d), and another methylene proton signals at δH 1.73 (2H, m) were observed. COSY correlation of the triplet methyl (H14) to a multiplet methylene (H13) gave an ethyl group, which was in turn connected to C5 by HMBC correlations from H13 and H14 to this carbon. Connectivity for C6/C2/C3/C4 was established by HMBC correlations from a singlet methyl H12 to C2 (δC 139.0), C3 (δC 172.3) and C4, and H7 and H11 to C2. Finally, HMBC correlations from geminally coupled methylene protons H4 to C5, C1 (δC 210.2), and C2 allowed the assembly of a cyclopentenone core. A hydroxy group was placed at C5 in consideration of the carbon chemical shift and the molecular formula to complete the planar structure of 6 (Fig. 3). Further exploration of the absolute configuration was hampered by the limited availability of the material and low reproducibility of production.

1H-1H COSY and key HMBC correlations for 6
2’-Oxosattabacin (7) was obtained as a pale yellow amorphous solid. The high-resolution ESITOFMS analysis gave an [M–H]– ion peak at m/z 203.1081, appropriate for a molecular formula of C13H16O2 (Δ-0.3 mmu, calcd for C13H15O2). The 13C NMR spectrum showed only ten carbon signals, suggesting the presence of symmetrical structure(s) in this molecule. In the 1H NMR spectrum, proton signals for a doublet methyl (δH 1.01, 6H), a methine (δH 2.27, 1H), and a methylene (δH 2.71, 2H) group were observed, and COSY-correlations connected these protons to give an isobutyl group (Fig. 4). Aromatic protons at δH 7.39 (1H), 7.40 (2H), and 7.84 (2H) were assigned to a phenyl group along with the carbons (δC 127.5, 128.6, and 130.2) directly bonding to these protons and an sp2 carbon (δC 134.2). The remaining proton signal at δH 6.45 was directly connected to a carbon at δC 113.2. These three fragments and two downfield oxygenated sp2 carbons (δC 197.6 and 147.1) were assembled into a single structure on the basis of HMBC correlations. A long-range correlation from the methylene H4′ to C3′ established the connectivity between C3′ and C4′. HMBC correlations from the olefinic proton H1′ to C2, C2′, and C3′ confirmed the carbon connectivity from C1 to C3′, thereby establishing the α,β-unsaturated ketone substituted by an isobutyl and a phenyl group. Finally, HMBC correlations from an exchangeable proton at δH 7.38 to C1′, C2′, and C3′ established the enol functionality, completing the structure of an enol form of 7 (Fig. 4). 1H and 13C NMR resonances and 2D NMR cross-peaks derived from the keto-form of 7 were also observed (Table 2, Figs. SI6–SI10). The ratio of two tautomeric isomers was ~4:1 in CDCl3. We next analyzed the configuration of the enol double bond. A NOESY correlation observed between H1′ and H4′ was suggestive of Z-configuration for C1′–C2′ double bond. In addition, the 3JCH coupling constant determined by the J-HMBC spectrum of 7 was 4.1Hz for a long-range coupling between H1′ and C3′, but this value was equivocal to undoubtedly assign the geometrical isomer (see Fig. S20 for typical examples of 3JCH values of a set of E- and Z-olefins). We thus synthesized two models (Z)-12 and (E)-12, in which the double-bond geometry was fixed as a methyl vinyl ether to compare the 3JCH values of both E- and Z-isomers (Scheme 1).

1H–1H COSY and key HMBC correlations for 7

Synthesis of model compounds (Z)- and (E)-12. Reagents and conditions: (a) i-Pr2NEt, PhCHO, n-Bu2BOTf, CH2Cl2 –78 to 0 °C, 14/other diastereomers = 3.6:1; (b) TESOTf, 2,6-lutidine, CH2Cl2, 0 °C to rt, 36% from 13; (c) LiBH4, Et2O, MeOH, 0 °C to rt, 91%.; (d) TEMPO, KBr, aq. NaOCl, NaHCO3, CH2Cl2 0 °C; (e) isobutylmagnesium chloride, THF, 0 °C to rt, dr = 20:1; (f) AZADOL, KBr, aq. NaOCl, NaHCO3; (g) TBAF/AcOH, THF, 0 °C to rt, 49% from 16; (h) PhSeH, ADDP, n-Bu3P, THF, rt; (i) aq. H2O2, NaHCO3,THF, 0 °C, 54% from 18; (j) MgCl2, Et3N, PhCHO, TMSCl, THF, rt, then TFA, MeOH, rt, 14’/other diastereomers = 1.8:1
To obtain (Z)-12 and (E)-12 with defined double-bond geometry, we utilized the well-established syn-elimination of selenoxides (Scheme 1). The synthesis of (Z)-12 began with the syn-selective Evans aldol reaction of known oxazolidinone 13 [14, 15] and benzaldehyde to give an inseparable 3.6:1 mixture of 14 and its diastereomers. Silylation of the mixture afforded 15 and the corresponding diastereomers, the latter of which were removed by SiO2 column chromatography. The oxazolidinone 15 was converted straightforwardly into 17 in three steps (reduction to form 16, its oxidation to an aldehyde, and the Grignard reaction). Oxidation of 17 [16] followed by TES deprotection afforded 18, which was treated with PhSeH under the Mitsunobu conditions [17] to give stereochemically inverted selenide 19 as a single diastereomer. Treatment of 19 with H2O2 followed by concomitant syn-elimination of the resulting selenoxide 20 provided (Z)-12 in a geometrically pure state. The other geometrical isomer (E)-12 was obtained by following the same sequence of reactions as employed for the synthesis of (Z)-12, except that the aldol reaction in the first step was conducted anti-selectively to afford 14′ [18]. The geometrical configurations of (Z)-12 and (E)-12 were corroborated by NOE correlations depicted in Scheme 1. As shown in Fig. 5, the 3JCH coupling constant between H7 and C9 was 3.1 Hz for (Z)-12 and 7.3 Hz for (E)-12, which strongly supported the Z-configuration for the enol form of 7 (3JCH, 4.1 Hz).

Determination of double-bond geometry of 7 based on the 3JCH coupling
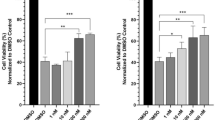
Compounds 6 and 7 were subjected to several biological testings. Preadipocyte differentiation into adipocytes is considered beneficial to insulin resistance and type 2 diabetes because matured adipocytes have an ability to secrete adiponectin, which promotes glucose uptake and improves insulin-sensitivity in type 2 diabetic patients [19]. Inducing potential of 6 and 7 toward murine ST-13 preadipocyte differentiation was thus evaluated at the concentrations of 10–40 μM by measuring the lipid droplet accumulation [20, 21]. 6 was weakly active (50% differentiation) at 40 μM, whereas 7 induced 69 and 85% differentiation at 10 and 20 μM, respectively. In addition, 6 and 7 were subjected to autophagy inducing activity assay. Autophagy is the cellular mechanism that eliminates unnecessary or malfunctional proteins by peptide degradation. Since impairment of autophagy is considered as a cause of an onset or development of cancer, neurodegenerative and inflammatory diseases, autophagy inducers are expected to prevent or treat these kinds of illnesses [22]. Autophagy inducing activity of 6 and 7 was assessed by using PC12 cells that express mCherry-GFP-LC3 tandem fluorescent construct (tf-LC3) [23]. According to the fluorescence imaging experiments, 7 significantly increased red puncta area at 1–10 μM after 24 h, indicating the activation of autophagy flux (Fig. 6). 6 was inactive in this assay. Both 6 and 7 showed no cytotoxicity against melanoma A375 cells at 10 μM and no appreciable activity in antimicrobial assay against Escherichia coli, Kocuria rhizophila, and Candida albicans.

Autophagy inducing activity of 6 and 7
In addition to 6 and 7, seven more benzenoid metabolites were isolated from the culture extract of strain NBRC 105916: sattabacin (2) [24], hazakacin (3) [9], 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid (4) [11], 3-phenylpropanoic acid (5), N-acetylanthranilic acid (8), N-propionylanthranilic acid (9), benzenepropanoic acid (10), and cinnamic acid (11) (Figs. 1, 2). Compounds 8 and 9 were previously found from a thermophilic bacterium Laceyella sacchari as its major metabolites in our laboratory [12]. Isolation of 2 and 3 from T. hazakensis SK20-1T was reported by Kuzuyama et al. [9]. They characterized an enzyme responsible for the synthesis of these acyloins. The enzyme catalyzed a carbon–carbon bond formation between two α-ketoacids, which coupled phenylpyruvic acid and 2-oxybutyric acid to yield an α-hydroxy-β-keto acid, a hypothetical intermediate of 3 (Fig. 7). α-Keto acids are known to be formed from the corresponding amino acids by oxidative deamination and the following hydrolysis: for example, l-phenylalanine is a precursor amino acid of phenylpyruvic acid, whereas ethylglycine, a plausible precursor amino acid of 2-ketobutyric acid, is quite uncommon in nature. To investigate the origin of 2-ketobutyric acid, incorporation of plausible 13C-labeled amino acid precursors was examined by feeding experiments. As expected, l-[4-13C]phenylalanine and [4-13C, 3-d2]-2-ketobutyric acid were incorporated into 3, labeling C1 and C5′, respectively (Fig. 8, Table 3). Then, we tested the feeding of labeled threonine because 2-oxobutyric acid is generated from l-threonine as an intermediate for isoleucine biosynthesis (Fig. 7) [25]. Actually, the feeding of l-[1,2,3,4-13C]threonine gave the labeling of C3′, C4′, and C5′ with high efficiency of incorporation (Fig. 7, Table 3). To the best of our knowledge, this is the first example of utilization of 2-oxobutyric acid as a building block of secondary metabolites.

Biosynthesis of hazakacin (3) in Thermosporothrix hazakensis

Incorporation of 13C-labeled precursors into hazakacin (3)
In conclusion, our chemical investigation on T. hazakensis NBRC 105916 led to the discovery of two structurally unique benzenoid-type secondary metabolites. Cyclopentenone, the core structure of ktedonoketone (6), is widely distributed in nature but those that have a phenyl substitution at the α-position is limited: only two examples, alternarienoic acid [26] and sydowins [27] are known both from fungi. 2′-Oxasattabacin (7) is a dehydro-congener of sattabacin (2) and is likely produced by the oxidation of 2. 7 is known as a product of chemical synthesis [28] or enzymatic reaction [29], but this is the first finding as a natural product. Many of the compounds characterized in this study are originated from amino acids, featuring the unique metabolism developed in the genus Thermosporothrix.
Experimental section
General experimental procedures
Optical rotation was measured using a JASCO DIP-3000 polarimeter. UV spectra were recorded on a Hitachi U-3210 spectrophotometer. IR spectra were measured on a Perkin Elmer Spectrum 100. NMR spectra were obtained on a Bruker AVANCE 400 or a Bruker AVANCE 500 spectrometer. HRESITOFMS were recorded on a Bruker microTOF focus. Cosmosil 75C18-PREP (Nakalai Tesque, Inc., 75 μm) was used for ODS column chromatography.
Microorganism
T. hazakensis NBRC 105916 was purchased from Biological Resource Center, National Institute of Technology and Evaluation, Chiba, Japan. The strain was maintained on a Bn-2 agar medium at 50 °C and transferred to a fresh medium every 2 weeks. Well-grown culture was also preserved at 4 °C.
Fermentation
T. hazakensis NBRC 105916 cultured on a Bn-2 medium consisting of soluble starch 0.5%, glucose 0.5%, meat extract (Kyokuto Pharmaceutical Industrial Co., Ltd) 0.1%, yeast extract (Difco Laboratories) 0.1%, NZ-case (Wako Chemicals USA, Inc.) 0.2%, NaCl 0.2%, CaCO3 0.1%, and agar 1.5% (pH 7.0) at 50 °C was inoculated into 500-ml Erlenmeyer flasks with baffles each containing 100 ml of the seed medium consisting of soluble starch 1.0%, glucose 0.5%, NZ-case 0.3%, yeast extract 0.2%, Tryptone (Difco Laboratories) 0.5%, K2HPO4 0.1%, MgSO4 7H2O 0.05%, and CaCO3 0.3% (pH 7.0). The flasks were placed on a rotary shaker (130 rpm) at 50 °C for 4 days. The seed culture (20 ml) was transferred into 2-l Erlenmeyer flasks with baffles each containing 500 ml of the A-11M production medium consisting of glucose 0.5%, glycerol 2.0%, soluble starch 2.0%, Pharmamedia (Traders Protein) 1.5%, yeast extract 0.3%, and Diaion HP-20 (Mitsubishi Chemical Co.) 1.0%. The pH of the medium was adjusted to 7.0 before sterilization. The inoculated flasks were placed on a rotary shaker (60 rpm) at 50 °C for 6 days.
Extraction and isolation
At the end of the fermentation period, 500 ml of 1-butanol were added to each flask, and the flasks were allowed to shake for 1 h. The mixture was centrifuged at 4000 rpm for 8 min, and the organic layer was separated from the aqueous layer containing the mycelium. Evaporation of the solvent gave ~5.67 g of extract from 6 l of culture. The crude extract was subjected to silica gel column chromatography with a step gradient of CHCl3/MeOH (1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1v/v). Fraction 3 (10:1) was concentrated to provide 1.45 g of brown oil, which was further purified by ODS column chromatography with a gradient of MeCN/0.1% HCO2H solution (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2 v/v), and MeOH. Fractions 3 (4:6), 4 (5:5), and 5 (6:4) were evaporated separately, and the resultant aqueous solution was extracted with EtOAc. After drying with anhydrous Na2SO4, the EtOAc layer was concentrated to give brown oil (Fr. 3: 134 mg; Fr. 4: 148 mg; Fr. 5: 254 mg). Fr. 4 was subjected to HPLC purification (Nakalai Tesque, Cosmosil C18-AR-II, 20 × 250 mm, 15 ml min−1, UV detection at 210 nm) with MeCN/0.1% HCO2H (20:80), followed by evaporation and extraction with EtOAc, to give benzenepropanoic acid (10, 6.5 mg, tR 18.7 min), hazakacin (3, 37.0 mg, tR 21.5 min), and ktedonoketone (6, 2.9 mg, tR 27.3 min). HPLC purification from Fr. 5 using the same column condition with MeCN/0.1% HCO2H (42:58) followed by evaporation and extraction with EtOAc gave sattabacin (2, 105.9 mg, tR 24.7 min). N-Acetylanthranilic acid (8, 1.3 mg, tR 13.1 min) and N-propionylanthranilic acid (9, 4.3 mg, tR 24.4 min) were obtained from Fr. 3 by HPLC purification (Nakalai Tesque, Cosmosil C18-AR-II, 10 × 250 mm, 4 ml min−1, UV detection at 210 nm) with MeCN/0.1% HCO2H (20:80), followed by evaporation and extraction with EtOAc. The silica gel fraction 2 (20:1) was evaporated to give 31.4 mg of yellow oil, which was then fractionated by LH-20 gel filtration (CH2Cl2–MeOH = 1:1) to yield 2′-oxosattabacin (7, 3.2 mg). Silica gel column fractions Fr. 6 (1:1) and Fr. 7 (0:1) were combined and concentrated in vacuo. The residue (1.90 g) was fractionated by ODS column chromatography in a similar manner as described above. Fraction 4 (5:5) was concentrated and extracted with EtOAc to give a brown solid (25 mg) which was purified by HPLC (Nakalai Tesque, Cosmosil C18-AR-II, 10 × 250 mm, 4 ml min−1, UV detection at 254 nm) with MeCN/0.1% HCO2H solution (33:67), followed by evaporation and extraction with EtOAc to give 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid (4, 4.3 mg, tR 17.1 min).
Ktedonoketone (6): pale yellow oil; \({[{\rm{\alpha}}]^{23}_{\mathrm{D}}}\)+ 12 (c 0.04, CHCl3); IR νmax 3378, 1703 cm−1; UV (MeOH) λmax (log ε) 202 (4.06), 223 (3.73), 300 (2.99); For 1H and 13C NMR data, see Table 1; HRESITOFMS [M+Na]+ 239.1043 (calcd for C14H16O2Na, 239.1044).
2’-Oxosattabacin (7): pale yellow amorphous; IR νmax 3375, 1666, 1634 cm−1; UV (MeOH) λmax (log ε) 233 (3.29), 309 (3.87); For 1H and 13C NMR data, see Table 2; HRESITOFMS [M–H]− 203.1081 (calcd for C13H15O2, 203.1078).
Feeding experiment
l-[1,2,3,4-13C]Threonine (Sigma-Aldrich), l-[4-13C]phenylalanine (Cambridge Isotope Laboratories, Inc.), and [4-13C, 3,3-d2]-2-ketobutyric acid sodium salt (Cambridge Isotope Laboratories, Inc.) were used for feeding experiments. In all cases, 13C-enriched carbon sources were dissolved in distilled water (4 mg ml−1). Each solution was sterilized by filtration through 0.2 -μm filter (Minisart), and 5 ml was fed to 500 ml of growing culture in a 2-l Erlenmeyer flask of T. hazakensis NBRC 105916. Feeding of each labeled precursor (20 mg per flask) was commenced 2 days after inoculation and continued four times at 24-h intervals. After a total of 144-h (6 days) incubation, the culture broth was extracted and 13C-labeled hazakacin (3) was isolated as described in the Extraction and isolation section.
Adipocyte differentiation assay
The assay was carried out according to the reported procedure [20, 21]. In brief, mouse-derived ST-13 preadipocytes were plated at 1 × 104 cells/ml in 24-well dishes containing 1 ml of a basal medium containing DME/F-12, 10% FBS, 100 units/ml penicillin, and 100 μg ml−1 streptomycin on day 1, and cultured at 37 °C in a humidified atmosphere containing 5% CO2. Moreover, the fresh basal medium containing the indicated concentrations of test compounds (or 20 nM rosiglitazone (positive control)) was replaced on day 2, 5, and 8. On day 12, the ST-13 cells were washed three times with phosphate-buffered saline, fixed with 10% formalin neutral buffer solution at room temperature for 10 min, and then washed with distilled water to remove formalin solution. Furthermore, the cells were rinsed with 60% isopropanol for 5 min, stained with 0.24 % oil red O at room temperature for 20 min, and then were photographed under a phase contrast microscope (x100 magnification) equipped a CCD camera (Leica Microsystems Japan, Tokyo, Japan).
Autophagy inducing assay
The assay was carried out according to the reported procedure [30]. In brief, autophagy induction was evaluated by fluorescence imaging with tandem fluorescent-tagged LC3 (mCherry-EGFP-LC3B, tf-LC3). Inducible tf-LC3 expressing clones was generated by transfection of pTRE2hyg/tf-LC3 plasmid vector to PC12/tet-on cells (631137, TaKaRa, Shiga, Japan). After 72 h differentiation with 100 ng ml−1 of NGF, PC12/tet-on/tf-LC3 cells were co-treated with 1 μg ml−1 of doxycycline (D9891, Sigma-Aldrich, St. Louis, MO, USA) and the indicated concentrations of test compounds. After 24 h, the cells were fixed with 3% paraformaldehyde in PBS for 15 min. Fluorescence imaging was performed with a confocal laser scanning microscope system (Fluoview FV1000, Olympus, Tokyo, Japan) equipped with a ×60 oil objective lens and an inverted microscope (IX81, Olympus). To quantify autophagy induction, mCherry-positive-GFP-negative puncta area was calculated using ImageJ software (http://rsb.info.nih.gov/ij/).
References
Bull AT, Asenjo JA. Microbiology of hyper-arid environments: recent insights from the Atacama Desert, Chile. Antonie Van Leeuwenhoek. 2013;103:1173–9.
Xu C, Sun X, Jin M, Zhang X. A novel benzoquinone compound isolated from deep-sea hydrothermal vent triggers apoptosis of tumor cells. Mar Drugs. 2017;15:E200.
Cavaletti L, et al. New lineage of filamentous, spore-forming, gram-positive bacteria from soil. Appl Environ Microbiol. 2006;72:4360–9.
Yabe S, Aiba Y, Sakai Y, Hazaka M, Yokota A. Thermosporothrix hazakensis gen. nov., sp. nov., isolated from compost, description of Thermosporotrichaceae fam. nov. within the class Ktedonobacteria Cavaletti et al. 2007 and emended description of the class Ktedonobacteria. Int J Syst Evol Microbiol. 2010;60:1794–801.
Yabe S, Aiba Y, Sakai Y, Hazaka M, Yokota A. Thermogemmatispora onikobensis gen. nov., sp. nov. and Thermogemmatispora foliorum sp. nov., isolated from fallen leaves on geothermal soils, and description of Thermogemmatisporaceae fam. nov. and Thermogemmatisporales ord. nov. within the class Ktedonobacteria. Int J Syst Evol Microbiol. 2011;61:903–10.
Yabe S, Aiba Y, Sakai Y, Hazaka M, Yokota A. Isolation of thermophilic bacteria from compost on antibiotic-supplemented medium. Microbiol Cult Coll (Jpn). 2011;27:73–78.
Chang YJ, et al. Non-contiguous finished genome sequence and contextual data of the filamentous soil bacterium Ktedonobacter racemifer type strain (SOS1-21T). Stand Genom Sci. 2011;5:97–111.
Park JS, et al. Identification and biosynthesis of new acyloins from thermophilic bacterium Thermosporothrix hazakensis SK20-1T. Chembiochem. 2014;15:527–32.
Yabe S, Sakai Y, Abe K, Yokota A. Diversity of Ktedonobacteria with actinomycetes-like morphology in terrestrial environments. Microbes Environ. 2017;32:61–70.
Vyssotski M, et al. A novel fatty acid, 12,17-dimethyloctadecanoic acid, from the extremophile Thermogemmatispora sp. (Strain T81). Lipids. 2012;47:601–11.
Park JS, Yabe S, Shin-ya K, Nishiyama M, Kuzuyama T. New 2-(1’H-indole-3’-carbonyl)-thiazoles derived from the thermophilic bacterium Thermosporothrix hazakensis SK20-1T. J Antibiot. 2015;68:60–62.
Akiyama H, et al. Metabolites from thermophilic bacteria I: N-propionylanthranilic acid a co-metabolite of the bacillamide class antibiotics and tryptophan metabolites with herbicidal activity from Laceyella sacchari. J Antibiot. 2014;67:795–8.
Akiyama H, et al. Linfuranones B and C, furnone-containing polyketides from a plant-associated Sphaerimonospora mesophila. J Nat Prod. 2018;81:1561–9.
Burke MD, Berger EM, Schreiber SL. A synthesis strategy yielding skeletally diverse small molecules combinatorially. J Am Chem Soc. 2004;126:14095–104.
Hunziker D, Wu N, Kenoshita K, Cane DE, Khosla C. Precursor directed biosynthesis of novel 6-deoxyerythronolide B analogs containing non-natural oxygen substituents and reactive functionalities. Tetrahedron Lett. 1999;40:635–8.
Shibuya M, et al. Practical preparation methods for highly active azaadamantane-nitroxyl-radical-type oxidation catalysts. Synthesis. 2011;11:3418–25.
Tsunoda T, Yamamiya Y, Itô S. 1,1’-(Azodicarbonyl)dipiperidine-tributylphosphine, a new reagent system for Mitsunobu reaction. Tetrahedron Lett. 1993;34:1639–42.
Evans DA, Tedrow JS, Shaw JT, Downey CW. Diastereoselective magnesium halide-catalyzed anti-aldol reactions of chiral N-acyloxazolidinones. J Am Chem Soc. 2002;124:392–3.
Kadowaki T, et al. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2016;116:1784–92.
Kunimasa K, et al. Identification of nobiletin, a polymethoxyflavonoid, as an enhancer of adiponectin secretion. Bioorg Med Chem Lett. 2009;19:2062–4.
Ikeda M, et al. Norlichexanthone isolated from fungus P16 promotes the secretion and expression of adiponectin in cultured ST-13 adipocytes. Med Chem. 2011;7:250–6.
Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24.
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26.
Lampis G, et al. Sattabacins and sattazolins: new biologically active compounds with antiviral properties extracted from a Bacillus sp. J Antibiot. 1995;48:967–72.
Grout DHG, et al. Stereochemistry of the conversions of L-threonine and D-threonine into 2-oxobutanoate by the L-threonine and D-threonine dehydratases of Serratia marcescens. Eur J Biochem. 1980;106:97–105.
Aly AH, et al. Cytotoxic metabolites from the fungal endophyte Alternaria sp. and their subsequent detection in its host plant Polygonum senegalense. J Nat Prod. 2008;71:972–80.
Teuscher F, et al. Two new cyclopentanoids from the endophytic fungus Aspergillus sydowii associated with the marine algae Acanthophora speciferas. Nat Prod Commun. 2006;1:927–33.
Dehmlow EV. Cyclopropenone chemistry. IV. Ring-opening reactions of 2-ethyl-3-phenylcyclopropenone. Justus Liebigs Ann Chem. 1969;729:64–68.
Kashiyama Y, Yoshikuni Y, Baker D, Siegel J. Recombinant microbial systems for converting polysaccharides into commodity products such as biofuels. PCT Int. Appl. 2009; WO 2009046370 A2 20090409.
Igarashi Y, et al. Nonthmicin, a polyether polyketide bearing a halogen-modified tetronate with neuroprotective and antiinvasive activity from Actinomadura sp. Org Lett. 2017;19:1406–9.
Acknowledgements
We thank to Dr. T. Okuda and Ms. Y. Sudoh for antimicrobial assay. This work was supported by JSPS KAKENHI Grant Number 24580156 to Y. I.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Igarashi, Y., Yamamoto, K., Ueno, C. et al. Ktedonoketone and 2’-oxosattabacin, benzenoid metabolites from a thermophilic bacterium Thermosporothrix hazakensis in the phylum Chloroflexi. J Antibiot 72, 653–660 (2019). https://doi.org/10.1038/s41429-019-0195-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0195-7
This article is cited by
-
Heterologous production of new lanthipeptides hazakensins A and B using a cryptic gene cluster of the thermophilic bacterium Thermosporothrix hazakensis
World Journal of Microbiology and Biotechnology (2023)
-
Complete NMR assignment and absolute configuration of k4610422, a norditerpenoid inhibitor of testosterone-5α-reductase originally from Streptosporangium: rediscovery from a thermophilic Actinomadura
The Journal of Antibiotics (2020)
