
Abstract
Covalently cross-linked rubber materials are widely used in elastic applications due to their excellent mechanical properties. However, the irreversibility of covalent cross-linking suffers from poor material recyclability. As a degradable polypeptide-based cross-linker for polybutadiene, poly(l-cysteine) (polyCys) was synthesized via papain-catalyzed chemoenzymatic polymerization. The resulting polyCys had intact thiol groups that cross-linked polybutadiene via the thiol-ene reaction. The cross-linking reaction of polybutadiene was performed in the presence of polyCys and a radical initiator and resulted in insoluble polybutadiene gel formation. Based on Raman spectroscopy analysis, the cross-linking reaction was confirmed by the consumption of thiol groups of polyCys. From the dynamic viscoelastic analyses of the cross-linked polybutadienes, the viscoelasticity drastically changed from that of raw polybutadiene, with the disappearance of the slow relaxation mode at low frequencies. The complete network formation was confirmed by E′ and E′′ showing the power law over the whole frequency range, as determined by the time-course experiments of the dynamic viscoelastic properties. The cross-linked polybutadiene was degradable by acid hydrolysis, resulting in the regeneration of soluble polybutadiene. Polypeptide cross-linked polybutadiene materials are promising candidates for the application of polybutadiene-based rubber materials with the requirements of both material integrity and reusability.
Similar content being viewed by others
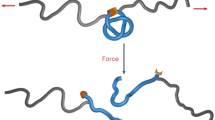


Introduction
Cross-linked rubber materials based on unsaturated hydrocarbon polymers, such as natural rubber, synthetic polyisoprene, and polybutadiene, are widely used in applications requiring elastic properties, such as tires, conveyor belts, and hose tubing [1]. Polybutadiene and polyisoprene rubbers are major commercial materials among a variety of petroleum-based synthetic rubbers. Additionally, natural rubber shows unique physical properties attributed to its chemical structure and ingredients, making natural rubber materials irreplaceable in some elastomer applications, such as tires [2,3,4,5]. Depending on the characteristics of matrix hydrocarbon polymers, cross-linked rubber materials exhibit a wide range of mechanical properties. For example, the chemical structure in the main chain of synthetic polybutadiene diverges into 1,2- and 1,4-units based on the polymerization mechanism, with cis and trans conformations existing for the 1,4-unit. Polybutadiene rubber with a high cis 1,4-unit content provides a viscoelasticity similar to natural rubber [1, 6], whereas 1,2-unit-rich polybutadiene is more crystalline and used for thermoplastic applications [7].
The cross-linking of these hydrocarbon polymers is generally accomplished by covalent bond formation with the carbon‒carbon double bonds on the polymer backbones. The vulcanization process is the most common industrial technique for cross-linking natural rubber by the reaction between double bonds of polyisoprene with sulfur at high temperatures [8,9,10]. Other chemical cross-linking systems have also been developed and industrialized [11,12,13,14]. Recent studies on cross-linked rubbers have focused on dynamic and reversible bonds for the cross-linking of rubbers via transient bond formation, such as dynamic covalent bonds, hydrogen bonds, or metal coordination [15,16,17,18]. Various types of dynamic cross-linking for rubber materials have been reported to date. Although these types of cross-linking provide reusability and easy processability, the transient bonds in theory provide weaker mechanical properties compared with covalently cross-linked rubber materials. If the hydrocarbon polymers can be cross-linked by a degradable cross-linker, the rubber materials are cross-linked by strong covalent bonds and recyclable by selective cleavage at the cross-linker units, as needed.
Polypeptides are composed of amino acids with a variety of side chains. Different features in the amino acid side chain in terms of size, hydrophobicity, and reactivity confer various functions and physical properties to the polypeptides. The peptide bond of polypeptides, which is occasionally susceptible to chemical or biological cleavage under certain conditions, provides structural integrity for materials, as observed in structural proteins [19]. Among amino acids, cysteine has a thiol group in the side chain and serves as a key residue for cross-linking in polypeptides and proteins by forming disulfide bonds. The reactive thiol group enables cysteine-containing polypeptides to react with polymer backbones via thiol-ene reactions. In general, the synthesis of cysteine-containing polypeptides suffers from a laborious synthetic process due to the protection/deprotection of the reactive thiol group. As an alternative facile synthetic method, we developed chemoenzymatic polymerization, which is the protease-catalyzed polymerization of amino acid ester monomers, to synthesize functional polypeptides with various types of side groups [20]. The substrate specificity of proteases used in chemoenzymatic polymerization provided a regioselective reaction, enabling the polymerization of amino acid monomers with intact reactive side groups, such as amine and thiol groups [21,22,23,24].
In this study, we aimed to cross-link unsaturated hydrocarbon polymers by cysteine-containing polypeptides that were possibly labile for chemical or biological degradation. The rubber materials cross-linked by polypeptides were reusable on demand by the selective cleavage of the peptide bonds in the cross-linker units. We selected 1,2-enriched polybutadiene with a high vinyl group (1,2-unit) content of ~65% as a model matrix polymer because of its higher reactivity compared with natural rubber (polyisoprene) and demonstrated that polybutadiene was effectively cross-linked with poly(l-cysteine) (polyCys) by a radical-mediated thiol-ene reaction.
Materials and method
Materials
Polybutadiene (number average molecular weight, Mn: ~9,000) was kindly donated by Bridgestone Corporation (Tokyo, Japan). l-cysteine ethyl ester (Cys-OEt) hydrochloride salt was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan) and used without purification. Papain (EC No. 3.4.22.2) was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) and was used as received. Its activity was approximately 0.5 U g−1, where one unit hydrolyzed 1 mmol of N-benzoyl-dl-arginine p-nitroanilide per minute at pH 7.5 and 25 °C. Deuterated trifluoroacetic acid (TFA-d) was purchased from Sigma–Aldrich (St. Louis, MO, USA). Deuterated chloroform (chloroform-d) was purchased from FUJIFILM Wako Pure Chemical Co. (Osaka, Japan). The other chemicals were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan) and used as received without purification unless otherwise noted.
Synthesis of polyCys by chemoenzymatic polymerization
Cys-OEt HCl salt (0.186 g, 1.00 mmol) and 1 M phosphate buffer (pH 8.0, 1.0 mL) were added to a 10 mL glass tube equipped with a stirring bar, and the mixture was stirred at 40 °C until the substrate was completely dissolved. Papain (50 mg) was then added to this solution. The final concentrations of monomer and papain were 1.0 M and 50 mg mL−1, respectively. The resulting mixture was stirred at 800 rpm and 40 °C for 2 h. After cooling to 25 °C, the precipitate was collected by centrifugation at 9000 rpm and 4 °C for 15 min. The crude product was washed with Milli-Q water and lyophilized to provide polyCys as a white solid. The yield was 90.1 mg (48%).
Cross-linking reaction of polybutadiene using polyCys
Polybutadiene (0.20 g) was dissolved in pyridine (1.0 mL). Subsequently, polyCys (1.25–5.0 mg) and 2,2’-azobis(isobutyronitrile) (AIBN) (12.0 mg, 30 equiv. to the vinyl group of polybutadiene) were added to the solution. The resulting mixture was stirred at 80 °C for 2 h. After the designated reaction time, pyridine was evaporated. The residual mass was then subjected to three washes with chloroform (5.0 mL) and methanol (5.0 mL) and subsequently dried under vacuum to produce cross-linked polybutadiene.
Hydrolysis of cross-linked polybutadiene under acidic conditions
Cross-linked polybutadiene (20 mg) was cut into small pieces (approximately 1 × 1 mm2) and placed in a mixed solvent of 1 M H2SO4 aq. and tetrahydrofuran (THF) in a volume ratio of 1/2. The mixture was vigorously stirred at 60 °C for 24 h. After the reaction, THF was removed by a rotary evaporator. The soluble fraction was extracted from the remaining aqueous dispersion using chloroform. The organic layer was filtered, concentrated by a rotary evaporator, and dried under vacuum. The resulting chloroform-soluble fraction was analyzed by 1H nuclear magnetic resonance (NMR) spectroscopy.
Analysis
Nuclear magnetic resonance (NMR) spectroscopy
The 1H NMR spectra were recorded on a Bruker DPX‐400 spectrometer (Karlsrube, Germany) at 25 °C at 400 MHz. Chloroform-d or TFA-d was used as the solvent, and tetramethylsilane (TMS) served as an internal standard.
Matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS)
MALDI-TOF mass spectrometric analysis was conducted using an ultrafleXtreme MALDI-TOF spectrophotometer (Bruker Daltonics, Billerica, MA) operating in linear positive ion mode. The sample was dissolved in TFA or water containing 0.1% TFA and mixed with a solution of α-cyano-4-hydroxycinnamic acid (CHCA) in acetonitrile.
Confocal Raman spectroscopy
All Raman spectra were obtained on a Jasco NRS-4500 laser Raman spectrometer (JASCO, Tokyo, Japan) with excitation at 532 nm. The cross-linked polybutadiene samples were placed on a microscope slide (SUPERFROST WHITE, S2441, Matsunami, Osaka, Japan). The laser output (UPLSAPO 100XO, Olympus, Tokyo, Japan) was focused on the sample, and standard Raman mapping at ×20 magnification was directly performed for each sample on the microscope slide with an acquisition time of 120 s at a light intensity of 50 mW μm−2 and a slit size of 100 μm × 8000 μm. For each sample, 200 spectra were collected over a 150 × 150 μm2 area, and the obtained spectra were averaged.
Wide-angle X-ray diffraction (WAXD) measurements
The synchrotron WAXD measurements of the cross-linked polybutadiene samples were performed on the BL05XU beamline (SPring-8, Harima, Japan) using an X-ray energy of 15 keV (wavelength: 0.82 Å). The bulk cross-linked polybutadiene was cut into a ring (3 and 7 mm inner and outer diameters, respectively), set on a stretching apparatus (see Fig. 4a) and placed on the beamline. The ring sample was irradiated with X-rays for 1 s at stretching ratios up to 400%. The two-dimensional (2D) diffraction pattern was obtained after subtracting the background pattern of air. The obtained 2D diffraction patterns were converted to one-dimensional (1D) azimuthal intensity profiles by azimuthal integration using Fit2D [25].
Dynamic viscoelastic measurements
Dynamic viscoelastic measurements were performed on the cross-linked polybutadiene samples by a stress-controlled rheometer (MCR301, Anton Paar, Graz, Austria) using a cone-and-plate fixture with a 10 mm diameter and a 2° cone angle. The samples were placed on the measuring plate, and the angular frequency (ω) dependence (0.1–100 rad s−1) of the storage modulus (G′) and loss modulus (G′′) was measured at 30 °C.
The time-course experiments for the dynamic viscoelastic property during the cross-linking reaction were performed by a strain-controlled rheometer (Rheogel E-4000, UBM Co., Kyoto, Japan) using a compression fixture. The sample mixtures of polybutadiene, polyCys, and AIBN were dispersed in toluene and cast on a parallel plate followed by drying overnight. We used toluene instead of pyridine for this experiment because toluene shows similar dispersibility of materials to pyridine, which has a malodorous odor. After the mixture was incubated at 80 °C for different times from 0 to 80 min with a 10 min interval, the frequency dependence (0.1–100 Hz) of the storage tensile modulus (E′) and loss tensile modulus (E′′) was measured at 30 °C under a nitrogen atmosphere. The oscillatory compressive strain amplitude was set at 0.03% and confirmed to be within the linear viscoelastic range before the measurements.
Results and discussion
Preparation of cross-linked polybutadiene using polyCys
Since cross-linked natural rubber materials are generally prepared by vulcanization in the presence of sulfur (S8) at high temperatures, we selected polyCys as a polypeptide-based cross-linking agent for the cross-linking reaction of polybutadiene. The thiol group of polyCys reacts with the double bonds of polybutadiene in the presence of a radical generator to form cross-linking points [26, 27]. PolyCys was synthesized by protease-mediated chemoenzymatic polymerization of l-cysteine ethyl ester according to the reported protocol [22]. Papain was used as an enzyme catalyst instead of the proteinase K previously used. The papain-catalyzed polymerization in a phosphate buffer produced polyCys as a white precipitate after 2 h of reaction. The characterization of polyCys was carried out using 1H NMR spectroscopy and MALDI-TOF MS spectrometry (Fig. 1). From the 1H NMR spectrum, the peaks were assignable to the backbone structure of polyCys and the C-terminal ethyl ester (Fig. 1a). The molecular weight of polyCys ranged from 600 to 1300, as determined by the MALDI-TOF MS spectrum (Fig. 1b). Two major series of peaks were assigned to polyCys with ethyl ester and hydrolyzed carboxylic acid termini. Notably, there was another series of small peaks at higher m/z, which corresponded to polyCys with one disulfide bond formation. The disulfide bond most likely formed during the purification step and the storage time. Most of the thiol groups of polyCys were predominantly kept intact during the papain-catalyzed polymerization because the obtained polyCys was completely soluble in TFA. The number average molecular weight calculated from the integral ratio in the 1H NMR spectrum was 8.1; however, this was slightly overestimated due to partial hydrolysis of the terminal ethyl ester, as confirmed by the MALDI-TOF MS spectrum. Collectively, polyCys was successfully synthesized by chemoenzymatic polymerization and directly used for the cross-linking reaction of polybutadiene.

1H NMR (a) in TFA-d and MALDI-TOF MS (b) spectra of polyCys obtained by papain-catalyzed chemoenzymatic polymerization
The cross-linking reaction of polybutadiene was carried out for a mixture of polybutadiene (Mn: ~9,000, 1,2-unit content: ~65% determined by 1H NMR, as shown in Fig. S1), polyCys, and AIBN as a radical initiator (Scheme 1). The mixture was dispersed in pyridine and heated at 80 °C for 2 h. The resulting cross-linked rubber material was washed with chloroform and methanol to remove unreacted polybutadiene and polyCys, respectively. We prepared three types of cross-linked polybutadiene (CL-PB) with different amounts of polyCys cross-linker (0.625, 1.25, and 2.5 wt% to polybutadiene), denoted as CL-PBx, where x is the amount of cross-linker (x = 0, 0.625, 1.25, and 2.5). Due to the low solubility of polyCys, a crosslinking reaction was not performed with a higher loading amount of polyCys than 2.5 wt%. The formulation conditions are summarized in Table 1. After the 2 h cross-linking reaction of polybutadiene, an insoluble gel was formed for all samples containing the polyCys cross-linker (Table 1, Runs 1–3). On the other hand, when polybutadiene was treated under the same conditions without polyCys, the resulting sample was still soluble in pyridine, and no cross-linked polybutadiene was obtained (Table 1, Run 4). This result indicates that heating with polyCys in the presence of the radical initiator successfully facilitated the cross-linking of polybutadiene via the thiol-ene reaction. However, prolonging the reaction time to 4 h resulted in the formation of an insoluble gel even without polyCys (Table 1, Run 5), indicating that radical-mediated cross-linking between polybutadiene main chains also occurred to some extent.

Cross-linking of polybutadiene via a thiol-ene reaction with polyCys
Characterization of the resultant CL-PB
The IR spectra of CL-PBs were almost identical to that of polybutadiene, and no information regarding the cross-linked structure was obtained (Fig. S2). Therefore, the chemical structure of CL-PBs was confirmed by Raman spectroscopy (Fig. 2, S3). The Raman spectrum of polyCys obtained by papain-catalyzed polymerization showed some characteristic peaks; the stretching vibration modes of S‒C (υS‒C) and S‒H (υS‒H) bonds at 690 and 2570 cm−1, respectively, were observed (Fig. 2a) [28]. The small peak at 500 cm−1 was assigned to the stretching vibration mode of the S‒S bond [29], which was consistent with the results from the MALDI-TOF MS analysis showing slight disulfide formation. As shown in Fig. 2c, the Raman spectra of CL-PBs were nearly the same as those of polybutadiene (Fig. 2b), with a small peak of the residual S‒H group of polyCys. When the cross-linking of polybutadiene with polyCys proceeded, additional S‒C bonds between polybutadiene and polyCys were formed, and the peak intensity of the S‒H bond decreased compared with that of the S‒C bond. We analyzed the peak area ratio of υS‒H to υS‒C in the Raman spectra after subtracting the polybutadiene-derived peak at approximately 675 cm−1 (Fig. 3) [28]. Compared with the peak area ratio of υS‒H to υS‒C for polyCys, all ratios of υS‒H to υS‒C for CL-PBs were smaller values. The cross-linking was heterogeneous due to the gradual formation of an insoluble gel during the reaction. This formation resulted in slightly scattered υS‒H/υS‒C values, which were inconsistent with the order of the polyCys amounts. However, the υS‒H/υS‒C values decreased for all CL-PBs, confirming that the thiol group was consumed by the radical-mediated thiol-ene reaction with polybutadiene to yield CL-PBs.

Raman spectra of (a) polyCys, (b) polybutadiene and (c) polybutadiene crosslinked with polyCys (CL-PB1.25)

Peak area ratio of υS-H (2570 cm−1) to υS-C (690 cm−1) in Raman spectra of polyCys and cross-linked polybutadienes
The cross-linking was further confirmed by WAXD analysis of CL-PB2.5 under stretch conditions. The ring sample of CL-PB2.5 was placed on a stretching apparatus and subjected to WAXD measurement with stretching at a certain ratio (Fig. 4a). The 2D WAXD profiles of CL-PB2.5 with and without stretching are shown in Fig. 4b, c. The CL-PB2.5 sample showed only an isotropic amorphous halo in the 2D WAXD profile. After stretching at λ = 4, the halo band became strong at the perpendicular angle to the stretching direction, as shown in Fig. 4c. The azimuthal intensity profiles of the halo band clearly showed an increase in the intensity at 90° and 270° after stretching (Fig. 4d). This result indicates that the cross-linked network structures oriented the polymer chains along the stretching direction [30, 31]. The ring sample was fractured at a relatively high stretching ratio of λ > 4, probably due to the heterogeneity of the cross-linking.

Ring sample of CL-PB2.5 for WAXD measurement during the stretching process (a) and the 2D WAXD profiles of CL-PB2.5 without stretching (b) and stretched at the stretching ratio λ = 4 (c). The stretching direction is horizontal to the images (0° and 180°). Azimuthal intensity profiles of the halo band for the sample with stretching ratios λ = 0 and 4 (d)
Dynamic viscoelastic measurements on CL-PBs
The viscoelastic properties of polybutadiene cross-linked by polyCys were investigated by dynamic viscoelastic measurements of CL-PBs. The bulk samples of CL-PBs were cut into a square plate, and the G′ and G′′ of the samples were measured at 30 °C and different frequencies (Fig. 5). As a control, a raw polybutadiene sample was also measured, and the G′ of the polybutadiene sample showed a typical viscoelastic relaxation at low frequencies, where the power law G′ ~ ω2 was confirmed [32]. In addition, G′′ also showed the power law G′′ ~ ω at low frequencies and a maxima with an uphill at high frequencies. Conversely, G’ surpassed G” at high frequencies, and the value of G’ at the high frequency limit effectively corresponded to that of the polybutadiene entanglement modulus [33]. The terminal flow behaviors of CL-PBs gradually shifted to lower frequencies with an increase in the polyCys content from 0.625 to 2.5 wt%, indicating that the relaxation was retarded by cross-linking. At the highest cross-linker amount of 2.5 wt%, G′ showed slight relaxation and no crossover with G′′, confirming network formation by cross-linking (Fig. S3) [33, 34]. The polybutadiene rubber cross-linked by vulcanization also showed similar G′ and G′′ behavior, supporting the cross-linking of polybutadiene by the reaction with polyCys [35]. Although CL-PB0.625 and CL-PB1.25 were insoluble gels after the cross-linking reaction, these samples still showed slight relaxation behavior at low frequencies. Moreover, the values of G’ at the high frequency limit were unchanged with the addition of PolyCys, indicating that the crosslink density was lower than the entanglement density. These findings indicate that the origin of the relaxation was potentially due to the occurrence of some disentanglements or the motion of dangling chains, along with the heterogeneity caused by gelation during the cross-linking reaction in pyridine.

Frequency dependence on (a) storage modulus (G′) and (b) loss modulus (G′′) of the cross-linked polybutadienes prepared with different polyCys contents at 80 °C for 2 h
The cross-linking of polybutadiene was further investigated by measuring the time course of the viscoelastic behavior of CL-PB0.625. The mixture of polybutadiene, polyCys, and AIBN was homogeneously dispersed in toluene, and the film sample was prepared by casting the dispersion followed by drying. The film sample was subjected to compressive dynamic viscoelastic analysis after each 10 min incubation up to 80 min at 80 °C under a nitrogen atmosphere. Figure 6 shows the frequency dependence of E′ and E′′ for CL-PB0.625 at different incubation times. Compared with the raw polybutadiene sample, the relaxation of the CL-PB0.625 sample for E′ and E′′ shifted to a lower frequency up to 60 min of incubation. A slow relaxation mode gradually dominated, especially for E′ at a frequency of ~ 1 Hz, indicating that cross-linking increased the molecular weight and branched structure. The relaxation at ~10 Hz was still present and likely corresponded to the relaxation of the dangling polybutadiene chains. After 70 min of incubation, both E′ and E′′ showed a power law over the entire frequency range (Fig. S4), corresponding to the gelation critical behavior reported by Winter and Chambon [36, 37]. For the 80 min incubation, a definite value of E′ larger than E′′ was observed in the low frequency range; thus, the cross-linking effectively and homogeneously proceeded compared with the reaction in pyridine solution (Fig. 5).

Frequency dependence of (a) storage tensile modulus (E′) and (b) loss tensile modulus (E′′) for CL-PB0.625 after incubation at 80 °C for different times. Raw polybutadiene without any cross-linking was used as a control
A time-course experiment was also performed for the sample mixture with a higher polyCys amount (CL-PB2.5, Fig. 7). Similarly, the slow relaxation mode gradually became pronounced at low frequencies for both E′ and E′′ as the incubation time increased and disappeared after a shorter incubation time of 30 min compared to CL-PB0.625. The spectra of E′ and E′′ with incubation times of 30 min and 40 min overlapped each other, indicating that a cross-linked network structure was obtained. The E′ value after 40 min of reaction for CL-PB2.5 was still lower than that of CL-PB0.625 after 70–80 min. Prolonging the reaction time would increase the E′ for CL-PB2.5, as revealed in Fig. 5 for the 2 h cross-linking reaction. Based on our results, more polyCys improved the cross-linking efficiency and shortened the incubation time for network formation.

Frequency dependence of (a) storage tensile modulus (E′) and (b) loss tensile modulus (E′′) for CL-PB2.5 after incubation at 80 °C for different times. Raw polybutadiene without any cross-linking was used as a control
Degradation of CL-PBs
The degradability of CL-PBs was investigated by chemical hydrolysis under acidic conditions. The polyCys cross-linker consisted of peptide bonds that were susceptible to hydrolysis by acid or base. CL-PB2.5 was treated with sulfuric acid in aqueous THF solution for 24 h (Scheme 2). As the reaction proceeded, the small piece of CL-PB2.5 became brittle. The chloroform-soluble part was extracted (36%) after the hydrolytic treatment, while almost no soluble part was obtained for pristine CL-PB2.5 by chloroform extraction. Notably, the cross-linked polybutadiene without polyCys was hardly degraded under the same conditions (only 4% of the soluble fraction was obtained by chloroform extraction). The residual CL-PB2.5 (64%) was insoluble in chloroform, indicating that the cross-linked structure was still maintained after hydrolysis under the current conditions. This occurred due to the hydrophobicity of the polybutadiene backbone, which impeded the penetration of aqueous media into the crosslinked network structure. In addition, undesired radical coupling between the double bonds partially occurred, resulting in nondegradable interchain cross-linking to some extent.

Hydrolytic cleavage of peptide bonds in the polyCys cross-linker by acidic treatment of CL-PBs
The 1H NMR spectrum of the obtained chloroform-soluble part is shown in Fig. 8. The 1H NMR spectrum showed characteristic signals similar to those of the original polybutadiene, with small signals assignable to the residual cysteine-related protons, as indicated by red arrows. The signals for the protons of the unsaturated double bonds were detected at 4.8–5.6 ppm, indicating that the reactive double bonds were still present after the cross-linking and hydrolysis cycle, and the recycled polybutadiene could be used to regenerate the cross-linked rubber material via a thiol-ene reaction.

a 1H NMR spectrum of the chloroform-soluble part of CL-PB2.5 after hydrolytic degradation in chloroform-d and (b) expanded spectrum at 2–4.5 ppm
Summary
We synthesized polyCys as a thiol-containing polypeptide-based cross-linker by the papain-catalyzed polymerization of l-cysteine ethyl ester. The obtained polyCys predominantly retained intact thiol groups. With the enzymatically synthesized polyCys as a cross-linker, we demonstrated that the cross-linking of 1,2-unit-rich polybutadiene was successfully performed by the thiol-ene reaction with polyCys in both solution and film states. The cross-linked structure was confirmed by both spectroscopic and dynamic viscoelastic experiments. Raman spectra showed that the thiol group of polyCys was effectively consumed by CL-PBs, indicating that the cross-linking of polybutadiene proceeded by the thiol-ene reaction with polyCys. From the dynamic viscoelastic analyses, the viscoelasticity of CL-PBs drastically changed from that of raw polybutadiene, with the disappearance of the slow relaxation mode at low frequencies. The complete network formation was confirmed by E′ and E′′ showing the power law over the entire frequency range, as determined by the time-course of the dynamic viscoelastic experiments. The polyCys cross-linker was cleaved by acid hydrolysis in aqueous media, enabling the regeneration of soluble polybutadiene from cross-linked polybutadiene. Polypeptide cross-linked polybutadiene materials are promising candidates for the application of polybutadiene-based rubber materials with the requirements of both material integrity and reusability. We envision further improvement in cross-linking/degradation efficiency for the peptide-based cross-linkers by designing more appropriate amino acid sequences instead of polyCys in future studies.
References
The Science and Technology of Rubber (Fourth Edition). (Academic Press, 2013).
Gimenez-Dejoz J, Tsunoda K, Fukushima Y, Numata K. Computational study of the interaction between natural rubber α-terminal groups and l-quebrachitol, one of the major components of natural rubber. Polym J. 2022;54:229–33.
Oouchi M, Ukawa J, Ishii Y, Maeda H. Structural analysis of the terminal groups in commercial Hevea natural rubber by 2D-NMR with DOSY filters and multiple-WET methods using ultrahigh-field NMR. Biomacromolecules. 2019;20:1394–1400.
Monadjemi SMA, Mcmahan CM, Cornish K. Effect of non-rubber constituents on Guayule and Hevea rubber intrinsic properties. J Res Updates Polym Sci. 2016;5:87–96.
Karino T, Ikeda Y, Yasuda Y, Kohjiya S, Shibayama M. Nonuniformity in natural rubber as revealed by small-angle neutron scattering, small-angle X-ray scattering, and atomic force microscopy. Biomacromolecules. 2007;8:693–9.
Kwag G. Ultra high cis polybutadiene by monomeric neodymium catalyst and its mechanical and dynamic properties. Macromol Res. 2010;18:533–8.
Kimura S-I, Shiraishi N, Yanagisawa S, Abe M. A new thermoplastic, 1,2-polybutadiene JSR RB- properties and applications. Polym Plast Technol Eng. 1975;5:83–105.
Shi F, Li X, Bai Y, Li L, Pu M, Liu L, et al. Mechanism of the zinc dithiocarbamate-activated rubber vulcanization process: a density functional theory study. ACS Appl Polym Mater. 2021;3:5188–96.
Dondi D, Buttafava A, Zeffiro A, Palamini C, Lostritto A, Giannini L, et al. The mechanisms of the sulphur-only and catalytic vulcanization of polybutadiene: An EPR and DFT study. Eur Polym J 2015;62:222–35.
Coran AY. Chapter 7 - Vulcanization. In: Mark JE, Erman B, Roland CM, editors. The Science and Technology of Rubber (Fourth Edition) Boston: Academic Press; 2013. p. 337-81.
Au-Duong A-N, Hsu Y-C, Chen K-L, Huang Y-S, Lai J-Y, Chiu Y-C. Intrinsically elastic and self-healing luminescent polyisoprene copolymers formed via covalent bonding and hydrogen bonding design. Polym J. 2022;54:1331–43.
Liu X, Zhou T, Liu Y, Zhang A, Yuan C, Zhang W. Cross-linking process of cis-polybutadiene rubber with peroxides studied by two-dimensional infrared correlation spectroscopy: a detailed tracking. RSC Adv. 2015;5:10231–42.
Bai J, Li H, Shi Z, Yin J. An eco-friendly scheme for the cross-linked polybutadiene elastomer via thiol–ene and Diels–Alder click chemistry. Macromolecules. 2015;48:3539–46.
Uhl FM, McKinney MA, Wilkie CA. Polybutadiene cross-linked with various diols — effect on thermal stability. Polym Degrad Stab 2000;70:417–24.
Verjans J, André A, Van Ruymbeke E, Hoogenboom R. Physically cross-linked polybutadiene by quadruple hydrogen bonding through side-chain incorporation of ureidopyrimidinone with branched alkyl side chains. Macromolecules. 2022;55:928–41.
Yamamoto K, Sogawa H, Takata T. Coordinative cross-linking of acrylonitrile–butadiene rubber with a macrocyclic dinuclear palladium complex. Polym J. 2021;53:565–71.
Salaeh S, Das A, Wießner S, Stapor M. Vitrimer-like material based on a biorenewable elastomer crosslinked with a dimeric fatty acid. Eur Polym J. 2021;151:110452.
Liu T, Zhao B, Zhang J. Recent development of repairable, malleable and recyclable thermosetting polymers through dynamic transesterification. Polymer. 2020;194:122392.
Numata K. How to define and study structural proteins as biopolymer materials. Polym J. 2020;52:1043–56.
Tsuchiya K, Numata K. Chemoenzymatic synthesis of polypeptides for use as functional and structural materials. Macromol Biosci. 2017;17:1700177.
Watanabe T, Terada K, Takemura S, Masunaga H, Tsuchiya K, Lamprou A, et al. Chemoenzymatic polymerization of l-serine ethyl Ester in aqueous media without side-group protection. ACS Polym Au. 2022;2:147–56.
Ma Y, Sato R, Li Z, Numata K. Chemoenzymatic synthesis of Oligo(l-cysteine) for use as a thermostable bio-based material. Macromol Biosci 2016;16:151–9.
Narai-Kanayama A, Hanaishi T, Aso K. α-Chymotrypsin-catalyzed synthesis of poly-l-cysteine in a frozen aqueous solution. J Biotechnol 2012;157:428–36.
Qin X, Xie W, Su Q, Du W, Gross RA. Protease-catalyzed oligomerization of l-lysine ethyl Ester in aqueous solution. ACS Catal. 2011;1:1022–34.
Tsuchiya K, Masunaga H, Numata K. Tensile reinforcement of silk films by the addition of telechelic-type polyalanine. Biomacromolecules. 2017;18:1002–9.
Lotti L, Coiai S, Ciardelli F, Galimberti M, Passaglia E. Thiol-Ene radical addition of L-cysteine derivatives to low molecular weight polybutadiene. Macromol Chem Phys 2009;210:1471–83.
Geng Y, Discher DE, Justynska J, Schlaad H. Grafting short peptides onto polybutadiene-block-poly(ethylene oxide): a platform for self-assembling hybrid amphiphiles. Angew Chem Int Ed. 2006;45:7578–81.
Bazylewski P, Divigalpitiya R, Fanchini G. In situ Raman spectroscopy distinguishes between reversible and irreversible thiol modifications in l-cysteine. RSC Adv. 2017;7:2964–70.
Zhang B, Crack JC, Subramanian S, Green J, Thomson AJ, Le Brun NE. et al. Reversible cycling between cysteine persulfide-ligated [2Fe-2S] and cysteine-ligated [4Fe-4S] clusters in the FNR regulatory protein. Proc. Nat. Acad. Sci.2012;109:15734
Liu C, Morimoto N, Jiang L, Kawahara S, Noritomi T, Yokoyama H, et al. Tough hydrogels with rapid self-reinforcement. Science. 2021;372:1078–81.
Toki S, Fujimaki T, Okuyama M. Strain-induced crystallization of natural rubber as detected real-time by wide-angle X-ray diffraction technique. Polymer. 2000;41:5423–9.
Vinogradov GV, Ya. Malkin A, Yanovskii YG, Borisenkova EK, Yarlykov BV, et al. Viscoelastic properties and flow of narrow distribution polybutadienes and polyisoprenes. J Polym Sci Part A-2: Polym Phys. 1972;10:1061–84.
Koike A, Nemoto N, Watanabe Y, Osaki K. Dynamic viscoelasticity and FT-IR measurements of end-crosslinking α,ω-dihydroxyl polybutadiene solutions near the Gel point in the gelation process. Polym J. 1996;28:942–50.
Kraus G, Rollmann KW. Dynamic viscoelastic behavior of polybutadiene networks. J Polym Sci Polym Symp. 1974;48:87–106.
Hess W, Vilgis TA, Winter HH. Dynamical critical behavior during chemical gelation and vulcanization. Macromolecules. 1988;21:2536–42.
Chambon F, Winter HH. Linear viscoelasticity at the gel point of a crosslinking PDMS with imbalanced stoichiometry. J Rheol. 1987;31:683–97.
Winter HH, Chambon F. Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J Rheol. 1986;30:367–82.
Acknowledgements
This work was supported by JST-Mirai Program Grant No. JPMJMI21EF, JST PRESTO Grant No. JPMJPR21N6, Grant-in-Aid for Transformative Research Areas (B) Grant No. JP20H05735, MEXT Data Creation and Utilization-type MaTerial R&D project, JST COI-NEXT, and JST CREST Grant No. JPMJCR2091. We would like to thank Bridgestone Corporation for kindly donating the polybutadiene samples.
Funding
Open access funding provided by The University of Tokyo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsuchiya, K., Terada, K., Tsuji, Y. et al. Cross-linking polybutadiene rubber via a thiol-ene reaction with polycysteine as a degradable cross-linker. Polym J 56, 391–400 (2024). https://doi.org/10.1038/s41428-023-00855-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-023-00855-9
